 Nicholas Weyandt
Nicholas Weyandt Shiva A. Aghdam
Shiva A. Aghdam Amanda M. V. Brown
Amanda M. V. Brown- Department of Biological Sciences, Texas Tech University, Lubbock, TX, United States
Wolbachia is a widespread endosymbiont of insects and filarial nematodes that profoundly influences host biology. Wolbachia has also been reported in rhizosphere hosts, where its diversity and function remain poorly characterized. The discovery that plant-parasitic nematodes (PPNs) host Wolbachia strains with unknown roles is of interest evolutionarily, ecologically, and for agriculture as a potential target for developing new biological controls. The goal of this study was to screen communities for PPN endosymbionts and analyze genes and genomic patterns that might indicate their role. Genome assemblies revealed 1 out of 16 sampled sites had nematode communities hosting a Wolbachia strain, designated wTex, that has highly diverged as one of the early supergroup L strains. Genome features, gene repertoires, and absence of known genes for cytoplasmic incompatibility, riboflavin, biotin, and other biosynthetic functions placed wTex between mutualist C + D strains and reproductive parasite A + B strains. Functional terms enriched in group L included protoporphyrinogen IX, thiamine, lysine, fatty acid, and cellular amino acid biosynthesis, while dN/dS analysis suggested the strongest purifying selection on arginine and lysine metabolism, and vitamin B6, heme, and zinc ion binding, suggesting these as candidate roles in PPN Wolbachia. Higher dN/dS pathways between group L, wPni from aphids, wFol from springtails, and wCfeT from cat fleas suggested distinct functional changes characterizing these early Wolbachia host transitions. PPN Wolbachia had several putative horizontally transferred genes, including a lysine biosynthesis operon like that of the mitochondrial symbiont Midichloria, a spirochete-like thiamine synthesis operon shared only with wCfeT, an ATP/ADP carrier important in Rickettsia, and a eukaryote-like gene that may mediate plant systemic acquired resistance through the lysine-to-pipecolic acid system. The Discovery of group L-like variants from global rhizosphere databases suggests diverse PPN Wolbachia strains remain to be discovered. These findings support the hypothesis of plant-specialization as key to shaping early Wolbachia evolution and present new functional hypotheses, demonstrating promise for future genomics-based rhizosphere screens.
Introduction
Host-associated microbes may play significant roles in the rhizosphere, but the complexity of these environments can make them challenging to study. Diverse microbiota has been detected in nematodes, whose communities number in the millions per m2 in the rhizosphere (Bongers and Ferris, 1999; Adam et al., 2014; Ferris and Tuomisto, 2015; Elhady et al., 2017; Zheng et al., 2020; Topalović and Vestergård, 2021). Such microbes may facilitate plant-feeding in rhizosphere organisms, as has been observed in above-ground plant-feeders, in which symbionts supplement essential nutrients (Moran et al., 2008; Bennett and Moran, 2013) or synthesize protective toxins or plant toxin-degrading enzymes (Cheng et al., 2013; Douglas, 2015; Gressel, 2018). Microbes could play significant roles in the biology of plant-parasitic nematodes (PPNs), which cause up to 25% of global crop yield loss and cost ∼$100 billion annually (Nicol et al., 2011). Recent findings suggest PPNs can harbor mutualist symbionts, like Xiphinematobacter (Verrucomicrobia) (Vandekerckhove et al., 2000; Brown et al., 2015) and Xiphinematincola (Burkholderiales) (Palomares-Rius et al., 2016, 2021), or symbionts with unknown phenotypes, including Cardinium (Bacteroidetes) (Noel and Atibalentja, 2006; Brown et al., 2018) and Wolbachia (Alphaproteobacteria) (Haegeman et al., 2009; Brown et al., 2016), but current studies on the diversity and roles of such microbes are limited.
Genus Wolbachia includes the most remarkable and widespread symbionts, which can act as obligate mutualists (Foster et al., 2005; Taylor et al., 2012; Nikoh et al., 2014) or parasites that manipulate host reproduction to promote transmission through the female germline using a wide array of phenotypes. Reproductive manipulating Wolbachia phenotypes include male-killing, parthenogenesis induction, feminization, or cytoplasmic incompatibility (CI), wherein infected males mating with uninfected females fail to produce progeny (Werren, 1997; Taylor and Hoerauf, 1999; Werren et al., 2008). Whereas Wolbachia is widespread in insects, its discovery in PPNs has significant implications, given the ecological importance of PPNs: if these Wolbachia are obligate mutualists like filarial nematode Wolbachia, then disruption of the symbiosis could reduce the PPN burden on plants, whereas if they are reproductive parasites conferring CI as in many insects, this phenotype could be harnessed for biocontrol analogous to Wolbachia-based control programs in mosquitoes (Caragata et al., 2016; Carey, 2018).
However, to date, the role of PPN-type Wolbachia remains ambiguous. For example, based on an analysis of the available genome derived from strain wPpe from the root-lesion nematode, Pratylenchus penetrans, comparative genomics could not fully resolve Wolbachia’s role (Brown et al., 2016). Phylogenomic analyses placed them at the root of the Wolbachia clade, supporting the early emergence of Wolbachia in ecdysozoan plant-diet specialists, perhaps supporting this diet. However, the genomic analysis suggested wPpe was devoid of most pathways typically seen in diet-supplementing Wolbachia such as riboflavin, biotin, thiamine (Brown et al., 2016). Alternatively, it may serve as a facultative nutritional mutualist synthesizing heme or mediating host iron homeostasis (Gill et al., 2014; Brown et al., 2016, 2018), or perhaps co-synthesizing fatty acids or methionine when in the presence of dual infection with Cardinium (Brown et al., 2018). Conversely, Wolbachia’s variable prevalence and correlation with female-biased populations in its PPN host P. penetrans (Wasala et al., 2019) hint at reproductive manipulation, while the high prevalence of its most closely related strain, wRad in the burrowing nematode, Radopholus similis (Haegeman et al., 2009), hints at possible mutualism. Unfortunately, to date, only one Wolbachia genome has been sequenced from a PPN (Brown et al., 2016).
Despite this limited genomic data and just three strains confirmed in three hosts (Haegeman et al., 2009; Brown et al., 2016; Brown, 2018; Wasala et al., 2019), PPN Wolbachia may be widespread. The evidence that PPN Wolbachia may be widespread derives from the evidence of Wolbachia-like horizontal gene transfers both recently and deeper in the nematode phylogeny (McNulty et al., 2010; Dunning Hotopp, 2011; Koutsovoulos et al., 2014; Husnik and McCutcheon, 2018), suggesting ancestral Wolbachia symbiosis even in lineages without current evidence of Wolbachia. Adding to this argument, current surveys of Wolbachia distribution may include false negatives, due to low titer infections, or PCR-screens with primers having mismatches to PPN-type Wolbachia (supergroup L) (Bordenstein et al., 2003; Augustinos et al., 2011), or inadvertent clearing of the symbiont resulting from routine antibiotic during culturing or processing of PPNs.
Therefore, to broaden our understanding of the early-branching Wolbachia in PPNs, this study screened rhizosphere nematode communities for Wolbachia. We performed phylogenomics and comparative genomics from metagenomic assembled genomes (MAGs), which has previously proven successful to gain insights into unculturable taxa (Brown et al., 2016; Scholz et al., 2020). We took advantage of genome skimming approaches (Denver et al., 2016; Myers et al., 2021) to simultaneously characterize PPNs, and also screened public databases. Mirroring recent work (Bennett et al., 2021; Myers et al., 2021), we also analyzed functional enrichment and signatures of selection to investigate Wolbachia’s role and transitions in function during evolution. The outcomes supported previous hypotheses about Wolbachia’s origin in plant-feeding nematodes and generated new hypotheses about specific core metabolic functions and horizontally transferred genes for symbiont-mediated nutrient pathways such as heme, lysine, and thiamine.
Materials and Methods
Field Sample Collection and Nematode Isolation From Soil and Roots
As part of a field survey seeking to uncover new nematode-associated endosymbionts, approximately 100–500 g of soil and roots were collected using a soil auger or serrated shovel, collecting from the top 15 cm of soil at a 30 cm to 1 m distance from the base of various plants at different sites (Supplementary Table 1). All handling and processing of samples were performed in compliance with USDA APHIS permits to A.M.V.B. Samples were kept cool (<10°C) until processed to isolate nematodes as described previously (Myers et al., 2021). Briefly, soil and roots were placed in Baermann funnels for 3–5 days, allowing nematodes to be collected. Nematodes were further separated from soil by sucrose flotation following (Jenkins, 1964) to remove remaining soil particles, and finally further washed to remove further contaminants by mixing in 40 mL distilled water, centrifuging, and discarding the supernatant.
DNA Extraction, PCR Pre-screening, and Illumina Library Preparation and Sequencing
To characterize nematode community and Wolbachia-like DNA, total nematode community DNA was isolated from each sample. After a brief examination under an inverted microscope, about 800 to 1,400 nematodes from each sample were exposed to five cycles of freeze-thaw to break cuticles. DNA was isolated using the Qiagen DNeasy Blood & Tissue Kit (Valencia, CA, United States) following the manufacturer’s directions. DNA quantity and quality were assessed on the Nanodrop spectrophotometer. An initial sample was prepared for sequencing and analysis without pre-screening as follows: approximately 0.6–1 μg of DNA was used for genomic library preparation with the QIAseq FX 96 DNA Library Kit (Valencia, CA, United States) following the manufacturer’s directions with fragmentation times and AMPure bead size selection steps optimized for 450–550 bp fragments. Library quality and quantity were assessed on the TapeStation 2200 (Agilent, USDA). Libraries were normalized and pooled before sequencing on Illumina HiSeq, with 150 PE cycles performed at Genewiz, Inc. (NJ). After positive results for a plant-parasitic nematode-associated Wolbachia strain were obtained from one sample (following bioinformatics analysis described below), subsequent samples from the same farm were pre-screened for the presence of Wolbachia prior to sequencing with custom primers designed based on published qPCR assay primers (Mee et al., 2015) that showed a high sensitivity for low-titer infections. However, as these primers had mismatches to 16S rRNA gene sequences from plant-parasitic nematode-associated Wolbachia strains, we modified the primer sequences to increase specificity to our targets using aligned sequences. The resulting new primers were: forward Wol-mee-F 5′-CTC ACA GAA AAA GTC CT-3′ and reverse Wol-mee-R 5′-CGC CTT TAC GCC CAA T-3′, with thermal cycle conditions: 95°C for 2 min, 35 cycles of 95°C for 30 s, 59°C for 30 s, 72°C for 1 min, and one cycle of 72°C for 10 min.
Draft Genome Assembly
To recover nematode and symbiont genomes, reads were de novo assembled (see details in Supplementary Material), then Wolbachia-like contigs were annotated. First, reads for each sample were filtered and trimmed using Trimmomatic version 0.38 (Bolger et al., 2014), and overlaps in paired reads were detected and merged in Pear version 9.11 (Zhang et al., 2014). Filtered paired reads and merged reads were de novo assembled with metaSPAdes version 3.13 (Bankevich et al., 2012; Nurk et al., 2017) using low kmers (25, 33, and 45). Assembly quality was assessed using Quast version 5.0.1 (Gurevich et al., 2013). Assemblies were screened for Wolbachia-like 16S rRNA using a two-step analysis with blastn in Blast + version 2.10.1 (Camacho et al., 2009) (−evalue 10) first to a custom database of Wolbachia 16S rRNA sequences, and then a second blastn to the complete NCBI nt database. Any samples with top blastn hits to PPN Wolbachia from this read-based blast were also considered “positive.” For PPN Wolbachia-positive samples, full genomes were extracted using similar two-step blastn searches, first to Wolbachia genome databases, then to the full nt database.
Based on the high sequence similarity among samples, and their origin from the same farm, we combined these samples for further analysis, to improve coverage and assembly quality. We used an iterative map-assemble approach using bwa version 1.17 (Li and Durbin, 2009) and a subtractive mapping approach as follows. First, we identified non-Wolbachia contigs in the initial assembles using both blastn results and a%GC filter using prinseq-lite.pl in the BRAbB software (Brankovics et al., 2016), then we used bwa mem to map each sample’s reads to this non-Wolbachia data specific to our samples, then we used samtools version 1.9 (Li et al., 2009) and custom scripts to extract unmapped (i.e., Wolbachia-enriched) reads. These enriched reads were concatenated for all samples and assembled in metaSPAdes with a kmers (25, 45, 65, and 99). The resulting assemblies were combined with Wolbachia-like contigs from the individual samples with the new strain. The process of bwa-based subtractive mapping was repeated from the original reads with this new, improved database. The new Wolbachia-enriched reads were again concatenated and de novo assembled again with metaSPAdes. This approach was repeated three times and stopped once the sum of the length of the resulting new strain Wolbachia-like contigs ceased to increase between cycles and inspecting genome contamination and completeness metrics using CheckM v1.0.18 (Parks et al., 2015) at intermediate steps.
The resulting contigs were assessed by several quality controls to reduce the likelihood of spurious bioinformatic contamination with non-Wolbachia data. To filter out contigs with potential short horizontally transferred Wolbachia-like DNA regions (HGTs), long contigs (>5,000 bp) were removed if coverage was >2 times the average coverage of the longest contigs, using coverage analysis in the pileup.sh in BBMap version 38.9 (Bushnell, 2014). To filter out possible HGTs, contigs were imported into Geneious Prime version 2020.0.4 (Biomatters, Ltd.) and inspected, with contigs >1,000 bp removed if GC content was below 24% or above 42%. Quality was assessed by annotating contigs using Prokka version 1.14.6 (Seemann, 2014) which uses Prodigal for ab initio gene prediction, HMMER3 for protein family profiles, BLAST+ for comparative annotation, Barrnap1 for rRNAs, Aragorn (Laslett and Canback, 2004) for tRNAs. The resulting genes were then analyzed with blastn to the nt database and with DIAMOND blastx to the full nr database to keep only contigs with the highest similarity to PPN Wolbachia. Finally, to check for regions of possible bifurcating misassembly due to mutational differences in the field sampled specimens, contigs were aligned and checked for blocks of near identity and synteny using Geneious Prime plugins ProgressiveMauve v1.1.1 (Darling et al., 2010) and LASTZ alignment v7.0.2 (Biomatters, Ltd.).
Gene and Pathway Annotation
The final assembly quality was analyzed in Quast version 5.0.1 (Gurevich et al., 2013) and re-annotated in Prokka. Completeness and contamination were assessed in CheckM and by evaluating the genome presence of housekeeping genes and tRNAs. Pathways were assessed by gene presence-absence comparisons with other Wolbachia and outgroups Anaplasmataceae using Roary version 3.13 (Page et al., 2015), and using ModelSEED version 2.6.1 (Seaver et al., 2021) which assesses metabolic models in the KEGG and MetaCyc databases. ModelSEED was run with “complete” supplemented in silico media.
Nematode Community Analysis and Nematode-Wolbachia Abundance Correlation
To assess possible nematode hosts for the Wolbachia from our sampled nematode communities, we compared 21 sampled nematode communities (Supplementary Table 1) that were extracted and sequenced as described above, as part of a broader study. All partial cytochrome oxidase I (COI) sequences with top blastn match to nematodes were extracted from our scaffolds using blastn and custom scripts. The relative abundance of nematodes in each sampled community was then estimated based on kmer coverage with the equation C = (CK.R)/(R − K + 1), where C is total coverage, CK is kmer coverage, K is the length of kmers, and R is read length and normalization to the total number of reads sequenced for each sample. To this matrix of COI hits, we added a column with the normalized coverage of Wolbachia 16S rRNA for the Wolbachia-positive samples. We then calculated and plotted Spearman rank correlation (rho) values and p-values using the R package Hmisc version 4.5-0 ‘rcorr’ which calculates a matrix of Spearman’s rho rank correlation coefficients for all pairs of columns for non-missing elements, using midranks for ties, with method “spearman,” order “hclust,” hclust.method “average” and plotting using ‘corrplot’. Multiple testing correction was performed using the Benjamini and Hochberg (1995) method in R with p.adjust “BH.”
Sequence Read Archive Screening
To survey publicly available sequencing projects for potential PPN Wolbachia-like sequences, we developed scripts that take input queries such as geographic locality, sample type, sequencing strategy, and access the NCBI Sequence Read Archive (SRA) data, then analyze downloaded datasets to screen for Wolbachia. Keywords include “rhizosphere,” “soil,” “grassland,” “forest,” “agriculture,” “nematode,” and strategies/platforms such as “MiSeq,” “HiSeq,” “Illumina,” “Amplicon,” or “wgs,” and combinations of these terms. Briefly, these scripts use the E-utilities public API from NCBI to obtain and analyze serially tabular sets of raw read data for SRA projects via concatenated SRR, ERR, and DRR run data, subjecting them to raw read 16S rRNA gene two-step blastn, as described above, using for the custom blastn all PPN Wolbachia, including the new Wolbachia 16S rRNA gene from the present study. For SRA experiments with positive results, reads were further assessed after adapter and quality trimming and filtering using Trimmomatic and merging using Pear as described above. The resulting Wolbachia-like sequences were then analyzed using phylogenetic analysis approaches described below.
Phylogenetic and Phylogenomic Analysis
To understand the evolutionary relationships among candidate PPN hosts, candidate PPN-type Wolbachia strains from SRA data mining, and the new Wolbachia strain compared to other strains, various phylogenetic analyses were performed. In most cases, the general approach involved first blastn searches of our sequences of interest to the nt database at NCBI to find ∼100–500 most closely related sequences. We then download and aligned these using either Mafft version 1.0.4 (Katoh and Standley, 2013) or Clustal Omega 1.2.3 (Sievers et al., 2011), and trimming and removing duplicates or highly similar sequences within the Geneious Prime version 2020.0.4 (Biomatters, Ltd.) suite, prior to phylogenetic analysis using both maximum likelihood (ML) phylogeny reconstruction was performed in RAxML version 4 (Stamatakis, 2014), assessing bootstrap support from 500 replicates and Bayesian phylogeny estimation with MrBayes version 2.2.4 (Huelsenbeck and Ronquist, 2001; Ronquist et al., 2012) with final phylogenetic trees visualized in FigTree version 1.4.42 with labels and color added in Adobe Illustrator. Specific phylogenetic analysis approaches are as follows.
For inferring relationships among candidate PPN hosts predicted from community correlation analyses (described above), partial nematode cytochrome oxidase 1 (COI) genes that were significantly correlated with Wolbachia-positive samples were aligned and analyzed with the ML GTR Gamma nucleotide model, with rate heterogeneity alpha estimated, and with rapid bootstrapping and search for the best-scoring ML tree (-f a -x 1) and Bayesian analysis with the GTR + G model with 4 categories, and Markov chain Monte Carlo settings of chain length 1,100,000, 4 heated chains, heated chain temp 0.2, subsampling frequency 200, Burn-in length 100,000, with random seed 31,569, and priors with unconstrained branch lengths GammaDir (1,0.1,1,1), checking for convergence with minESS > 200.
For inferring relationships among candidate PPN-type Wolbachia strains from SRA data mining, an initial set of thousands of trimmed and merged reads with top blastn similarity to PPN-type Wolbachia were aligned to reference 16S rRNA genes, then identical sequences were removed. Preliminary phylogenetic analyses in FastTree v2.1.11 (Price et al., 2010) with the GTR model were used to generate a preliminary tree to remove large numbers of sequences from the alignment that displayed exceptionally long branches, similar to the distance separating Ehrlichia/Anaplasma and Wolbachia clades, on the basis that these sequences may reflect either non-Wolbachia alphaproteobacterial, or possibly degrading 16S rRNA pseudogenes or degrading horizontally transferred 16S rRNA fragments. The resulting subset of SRA sequences was analyzed in three separate alignments for the sub-region of the 16S rRNA gene in which they occur, and then together in a larger alignment to include all sequences together. RaxML and MrBayes phylogenies were reconstructed as described above.
For inferring relationships among the new Wolbachia strain and other Wolbachia strains, we focused on three sets of analyses: two which included strain wRad which was the only other strain to date from a PPN, and one which included a larger number of genes even though wRad could not be included. For strain wRad, only 3 gene regions have been sequenced. The first analysis was able to include additional early-branching Wolbachia isolates for which only the 16S rRNA gene sequence was available, and this analysis included additional outgroups Candidatus Neowolbachia serbia and Candidatus Mesenet longicola (GenBank Accessions MH618374.1 and BNGT01000041.1, respectively) and related bacteria from Harpalus pennsylvanicus, as well as additional Rickettsiales and outgroup alphaproteobacterial. The second analysis included more gene regions available for strain wRad, including 16S rRNA, partial CTP synthase and ftsZ, and partial groES and groEL (GenBank Accessions EU833482.1, EU833483.1 EU833484.1, respectively) and corresponding gene regions for other Wolbachia and outgroups with gene regions with blastn matches to these from the NCBI nt and genome databases. Once aligned, the sequence block was concatenated for strains with all regions covered, and the final alignment was stripped of sites with gaps or ambiguities. We produced additional reduced-length alignments from this full gap-stripped alignment to test for potential long-branch attraction artifacts. These reduced alignments were generated by successively stripping sites with the highest evolutionary rates identified using TIGER v2.0 (Cummins and McInerney, 2011). Phylogenetic analyses on resulting blocks were performed as described above.
For a more robust phylogenomic analysis of PPN-type Wolbachia with their outgroups, we prepared an alignment of a larger number of core genes from full Wolbachia and outgroup genomes downloaded from NCBI. We used Roary to generate a codon-based alignment (core_gene_alignment.aln) block for analysis. As for the previous analyses, to control for potential alignment artifacts, we removed all positions with ambiguities or gaps and performed TIGER analysis to create additional shorter alignments with high evolutionary rate sites progressively removed, to potentially reduce the effects of long-branch attraction. ML and Bayesian analyses were performed as described above for each alternative alignment. Finally, using the initial codon-based alignment, we performed in silico translation, removed gaps and ambiguities, and performed ML analysis of amino acid sequences was performed with RaxML using the PROTGAMMAGTR substitution model with empirical base frequencies and 500 bootstrap replicates.
Gene Repertoire Comparisons via Gene Ontology Enrichment Analysis
To evaluate gene repertoire differences among Wolbachia strains and outgroups and analyze functional gene ontology (GO) enrichment, we first performed gene annotation on our new Wolbachia strain assembly as well as 200 other draft Anaplasmataceae genomes from NCBI using Prokka with identical parameters for all assemblies. We then created ortholog sets using Roary v3.13.0 (Page et al., 2015) which performed blastp on gff files from Prokka, with parameters -e for codon-aware alignment in PRANK (Löytynoja, 2014) and -i 60 to allow for detection of distant orthologs with outgroups. Initial Roary results suggested some draft genomes were potentially incomplete (too few genes) or contaminated (too many genes), so this genome set was reduced to the best genomes, comprising 93 draft genomes from Wolbachia strains plus 9 outgroup draft genomes. Roary gene clusters without clear gene annotations, listed as “group_#,” were re-examined and if possible assigned gene names by cross-referencing Prokka gene calls to gene and GO annotations for Wolbachia strains downloaded from UniProtKB database and other databases (MetaCyc, KEGG). These databases were also used to create a master GO annotation file for Anaplasmataceae for downstream enrichment analyses. Pangenome and core genome comparisons based on Roary gene_presence_absence.csv outputs were depicted with the online Venn drawing tool http://bioinformatics.psb.ugent.be/webtools/Venn/. Functional GO enrichment was assessed using topGO version 2.4.0 (Alexa and Rahnenfuhrer, 2020) which assesses GO-term graph topology ad creates test statistics using the algorithm ‘weight01.fisher’ which returns corrected p-values not affected by multiple testing. TopGO was implemented in R using the script aip_topgo_usage.consider_universe.R3 for multiple gene subsets (depicted in Venn diagrams) using ‘diff’ and ‘universe’ gene sets.
Analysis of Functional Enrichment for Gene Classes With Different dN/dS
To assess genes and gene regions that may be important functionally, through signatures of increased or decreased purifying or directional selection, we applied an analysis approach that assessed pairwise dN/dS followed by GO enrichment tests on various high and low dN/dS gene sets. These analyses involved first generating new pairwise codon-based nucleotide alignments of orthologs generated in Roary, for each pair of Wolbachia strains. We then generated sliding window blocks using the function split.java in KaKs Calculator 2.0 (Zhang et al., 2006) to create 1,200 bp length windows with overlaps of 600 bp, preserving codon positions. We then used KaKs Calculator to assess dN/dS on all windows, specifying genetic code 11. This program assesses Ka/Ks (or dN/dS) while controlling for multiple substitutions per site and using a maximum likelihood framework for model selection and AICc for model averaging (Zhang et al., 2006). In addition to multiple substitution corrections performed by this software, we also set a maximum Ks cutoff for our output data of 2 for Wolbachia pairs in the major supergroups, and 2.5 for Wolbachia in the PPN supergroup. Annotated genes were matched and counted as within windows if they occurred across at least 300 bp of a given window. Output dN/dS values were grouped into subsets for topGO functional enrichment analysis, partitioning genes into sets with values into top 10%, top 25%, the bottom 10%, and bottom 25% of each pairwise comparison.
Results
Nematode Communities Were Screened and Found Positive for Wolbachia
Screening for PPN-type Wolbachia was initially performed on 16 samples (Supplementary Table 1) with the initial screening of raw reads showing 10 out of 16 samples with reads mapping to the Wolbachia 16S rRNA gene. However, blastn analysis of these reads showed that most of these read hits were more similar to non-PPN Wolbachia strains than to PPN strains, with only one sample (P3-11) showing high similarity to wPpe from the plant-parasitic nematode P. penetrans and wRad Wolbachia from R. similis. This sample was from a fruit tree farm in Los Fresnos, Texas (26.1585 N 97.3844 W) consisting of pooled soils from the following fruit trees: mango (Mangifera indica; Sapindales: Anacardiaceae), guava (Psidium guajava; Myrtales: Myrtaceae), pomello (Citrus maxima; Sapindales: Rutaceae), sugar-apple (Annona squamosa; Magnoliales: Annonaceae), and sapodilla (Manilkara zapota; Ericales: Sapotaceae). After the discovery of this initial positive result, separate samples were collected from 14 individual fruit trees at the same farm, from which the following five fruit tree samples were found to be PCR-positive and were processed for community shotgun sequencing: sugar-apple, avocado (Persea americana; Laurales: Lauraceae), plantain (Musa × paradisiaca AAB; Zingiberales: Musaceae), guava, and “Rosetta fruit” (Malus pumila “Niedzwetzkyana”; Rosales: Rosaceae) (Supplementary Table 1).
Genome Assembly and General Features of Wolbachia Strain wTex
Based on initial alignments of contigs from six Wolbachia-positive samples which originated from the same farm, inter-sample divergence was found to be low, therefore, to increase coverage, final genome assembly was performed for the six samples with reads pooled together. The final assembly (NCBI GenBank accession JAIXMJ000000000) of the new Wolbachia strain, hereafter, designated wTex (named for its location in southern Texas), consisted of 192 scaffolds with a total length of 1,013,022 bp, maximum scaffold length of 57,862 bp, N50 of 10,082, with 33.49% GC, coding density 0.809, with 989 predicted genes, and a full set of rRNA and tRNA genes (3 and 38, respectively), and average coverage of 15.96X (Supplementary Table 2). CheckM-based genome completeness was 84.37% based on 368 markers from 63 Rickettsiales genomes, with a contamination score of 0.64 CheckM. Prior to this final assembly, during iterative steps to improve the assembly CheckM completeness scores decreased as contamination scores decreased. Comparative genome features across similar Wolbachia strains are shown in Table 1. Among the predicted genes, 398 (40.2%) had no known function. GC content (Figure 1) was intermediate between that of clades C and D Wolbachia strains from filarial nematodes and clades A and B from arthropods, whereas assembly length was shorter than that of the majority of clades A and B Wolbachia strains and was more similar to that of clades C and D Wolbachia.
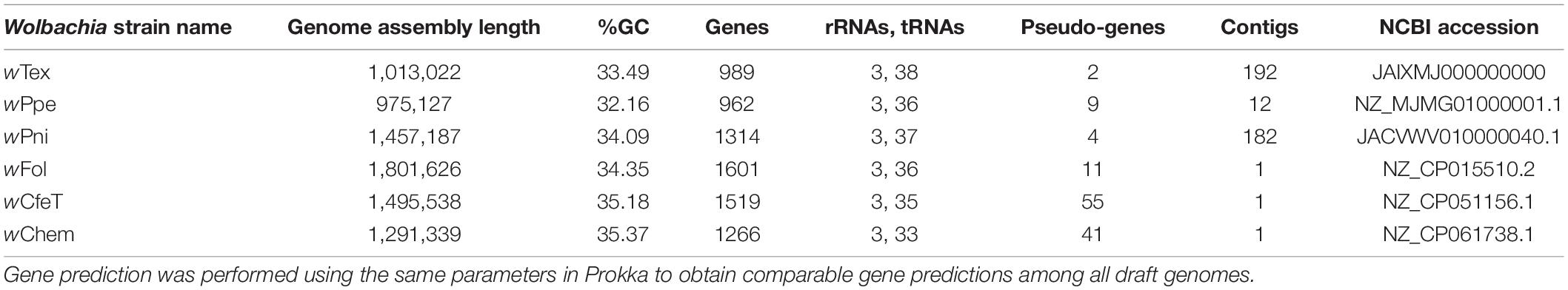
Table 1. Comparative gene and genome features for whole-genome shotgun assemblies for the new Wolbachia strain, designated wTex, and its closest relatives, wPpe from the plant-parasitic nematode Pratylenchus penetrans, wPni from the banana aphid Pentalonia nigronervosa, wFol from the springtail Folsomia candida, wCfeT from the cat flea Ctenocephalides felis, wChem from the bedbug Cimex hemipterus.
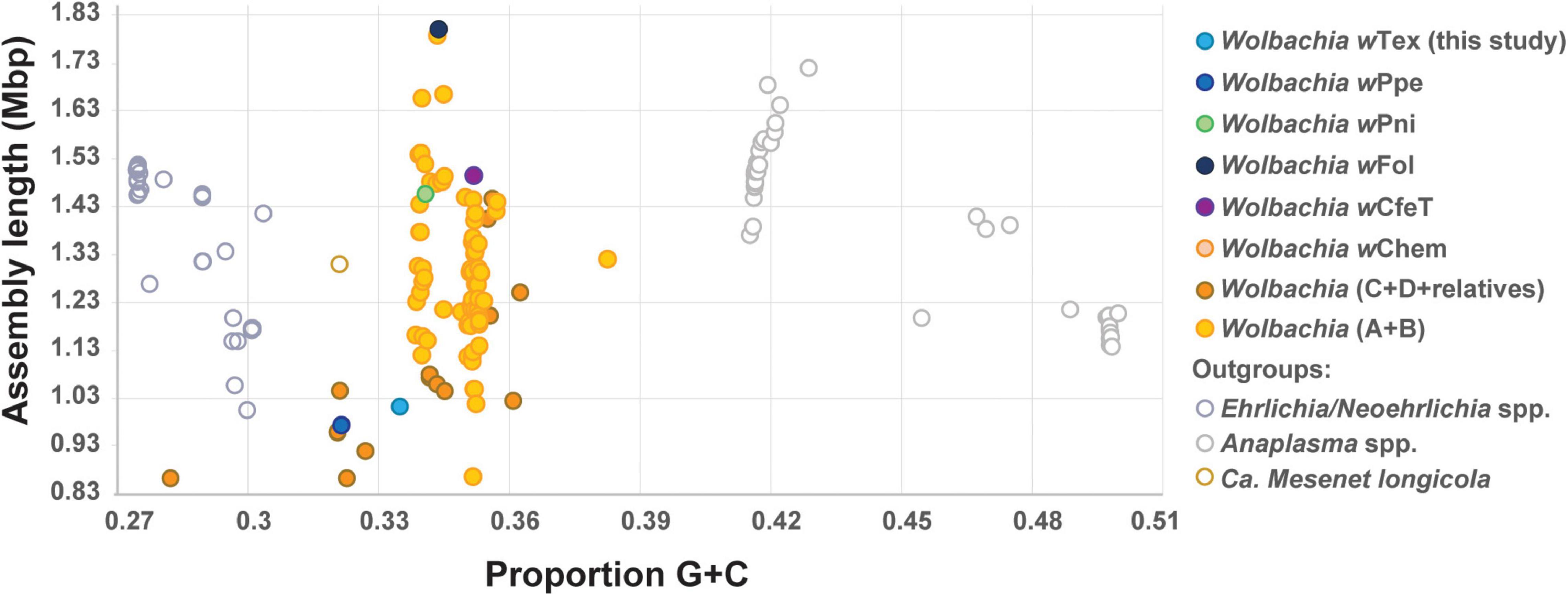
Figure 1. Plot of estimated genome sizes (total assembly lengths) versus proportion G + C content for Anaplasmataceae including Wolbachia and outgroups, showing PPN-type strains wTex and wPpe and early-branching clades wPni and wFol, and others.
Correlation Between Nematodes and Wolbachia-Positive Samples and Cytochrome Oxidase I Phylogeny of Candidate wTex Hosts
Correlation analyses (Supplementary Figure 1) showed Wolbachia wTex was positively associated with nematode hits to the partial COI gene of Heteroderidae sp. CD2526 (GenBank accession MK033155.1) (rho 0.659044735, BH-corrected p-value 0.02339631) and Ptycholaimellus sp. M1 (GenBank accession KX951909.1) (rho 0.713184097, BH-corrected p-value 0.006335523), however, hits matching the latter were very short and these scaffolds were at very low coverage. Scaffolds identified as matches to the COI of Heteroderidae sp. CD2526 were generally longer > than 1,000 bp with longer blast matches and were therefore extracted for further analysis. This produced 9 scaffolds with similarities to this COI hit. Similar sequences from GenBank were downloaded and aligned and analyzed by ML and Bayesian methods, which showed 7 sequences clustered with support in a clade together with Helicotylenchus spp. while 2 sequences clustered in a sister-clade with Rotylenchus spp. (Supplementary Figures 2, 3).
Phylogenetic Analysis of wTex With Other Wolbachia Strains and Outgroups
Phylogenetic analyses for the 16S rRNA gene alone (Supplementary Figure 4) which included the broadest array of outgroups and additional early-branching Wolbachia in this study and an alignment block length of 1,573 nucleotide positions, resulted in a strongly supported clade for PPN Wolbachia strains wTex, wPpe, and wRad, in a basal position in the Wolbachia phylogeny. The closest early-branding sister taxa to this PPN Wolbachia clade included a well-supported cluster from various aphids including Pentalonia nigronervosa wPni-like strains (accessions NZ_JACVWV01000005.1, KJ786949.1, KJ786950.1) along with strains from the conifer aphid Cinara cedri (AY620430.1) and the trunk-feeding aphid Stomaphis sinisalicis (KF751211.1), and strains wBta from the whitefly Bemisia tabaci (KF454771.1) and wBry from the spider mite Bryobia spec. V (EU499316.1). The largest sequence difference in the 16S rRNA region among PPN-type Wolbachia strains was 4.211%, whereas the average difference between the PPN-clade and wPni-like strains was 3.934%. A separate early-branching sister clade consisted of closely related isolates of a Wolbachia from the fungal-feeding mold mite, Tyrophagus putrescentiae (Supplementary Figure 4).
Phylogenetic analyses for the concatenation of 3 gene regions (16S rRNA, ftsZ, and groEL) to better resolve the phylogeny including all three PPN-type strains wTex, wPpe, and wRad resulted in a strongly supported clade for PPN Wolbachia strains (Figure 2). The supported clade was obtained for both ML and Bayesian analyses, which produced similar tree topologies, and for all alternative alignments including the full sequence alignment, the gap-stripped alignment, and alignments with progressive stripping of high evolutionary rate sites resulting in alignment lengths of 2,368 to 4,674 nucleotide positions. Bootstrap support for the PPN Wolbachia clade was 85–100%, with support of 100% for the majority of the alternate alignments. The PPN Wolbachia clade formed a strongly supported earliest branch for the genus Wolbachia (supergroup L in Figure 2) for alignments with full data, gap-stripped data, and data with high evolutionary rate sites stripped that included over 68% of the original alignment length, however, progressive stripping of such high-rate sites placed strain wPni at the root of the tree with or without >50% bootstrap support and reduced support for other basal branches.
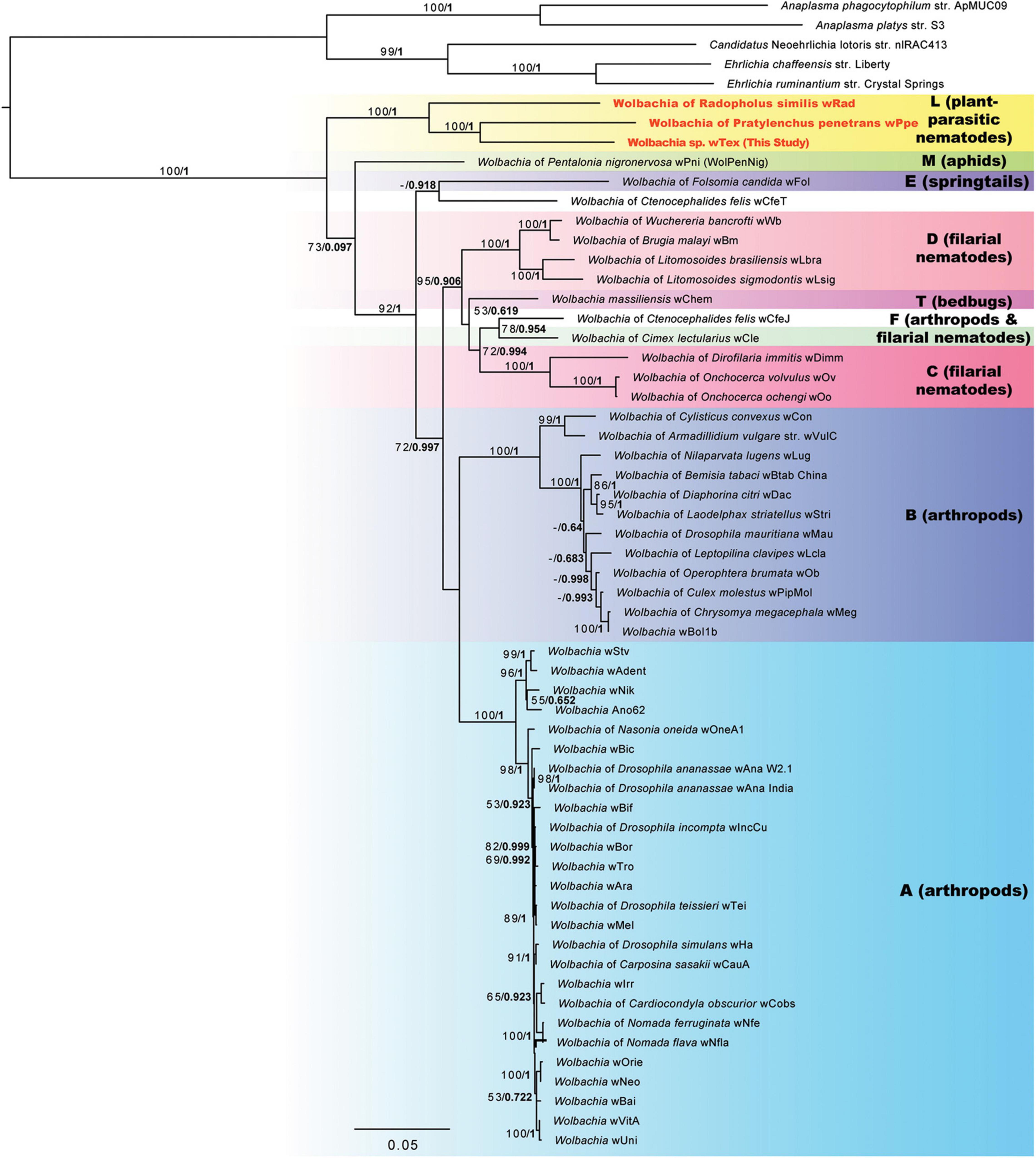
Figure 2. Phylogeny of three gene regions (16S rRNA, CTP synthase/ftsZ, and groES/groEL) for Wolbachia and outgroups based on 3,656 aligned positions of the concatenated nucleotides with gaps and ambiguities and the most variable 1/10th of positions removed. Maximum likelihood phylogeny reconstruction was performed in RAxML GTR + Gamma with bootstrap support >50% from 500 replicates shown on branches. Most supported nodes were obtained with high support in Bayesian 50% majority rule in MrBayes with GTR + G with 4 rate categories model. Bayesian posteriors are shown next to bootstrap values, in bold. Sequences obtained in this study are indicated in bold orange font.
Phylogenomic analyses of 100 genome-wide protein-coding genes (orthologs from Roary), resulted in similar results to the 3-region results (above), producing a strongly supported clade for PPN Wolbachia strains wTex and wPpe (Figure 3). ML bootstrap support and Bayesian posterior probabilities were largely similar for these analyses, showing support for this clade of 100% bootstrap and posterior of 1. These results were consistent for alternative alignments including the full sequence alignment, the gap-stripped alignment, an alignment with 3rd codon positions removed, a translated alignment, and alignments with progressive stripping of high evolutionary rate sites resulting in alignment lengths of 17,382 to 87,628 nucleotide positions. The exception to this high support was alignments stripped to less than 20% of the original positions, in which there was very little support for any Wolbachia clade. As with the 3-region phylogenetic analysis, the 100-gene phylogenetic analyses produced a strongly supported earliest branch position for the PPN Wolbachia clade (supergroup L in Figure 3) in some, but not all alternative alignments. For example, for alignments with full data, full data with gaps and ambiguities stripped, and full data with 3rd codon positions stripped, there was 100% bootstrap support for the PPN Wolbachia clade forming the earliest branch for Wolbachia. However, for alignments with high evolutionary rate sites stripped such that the alignment was 25–43% of its original length, the earliest branch of genus Wolbachia varied, sometimes placing wPni at the root of the tree just before PPN Wolbachia, and sometimes placing strains wFol and wCfeT as sisters to PPN Wolbachia, together forming the earliest branch. However, one alignment with high-rate sites stripped, with 22% of the original length, produced 91% bootstrap support for PPN Wolbachia at the root of the genus. Phylogenies based on amino acid alignments, however, also placed strain wPni at the root with 82% bootstrap support.
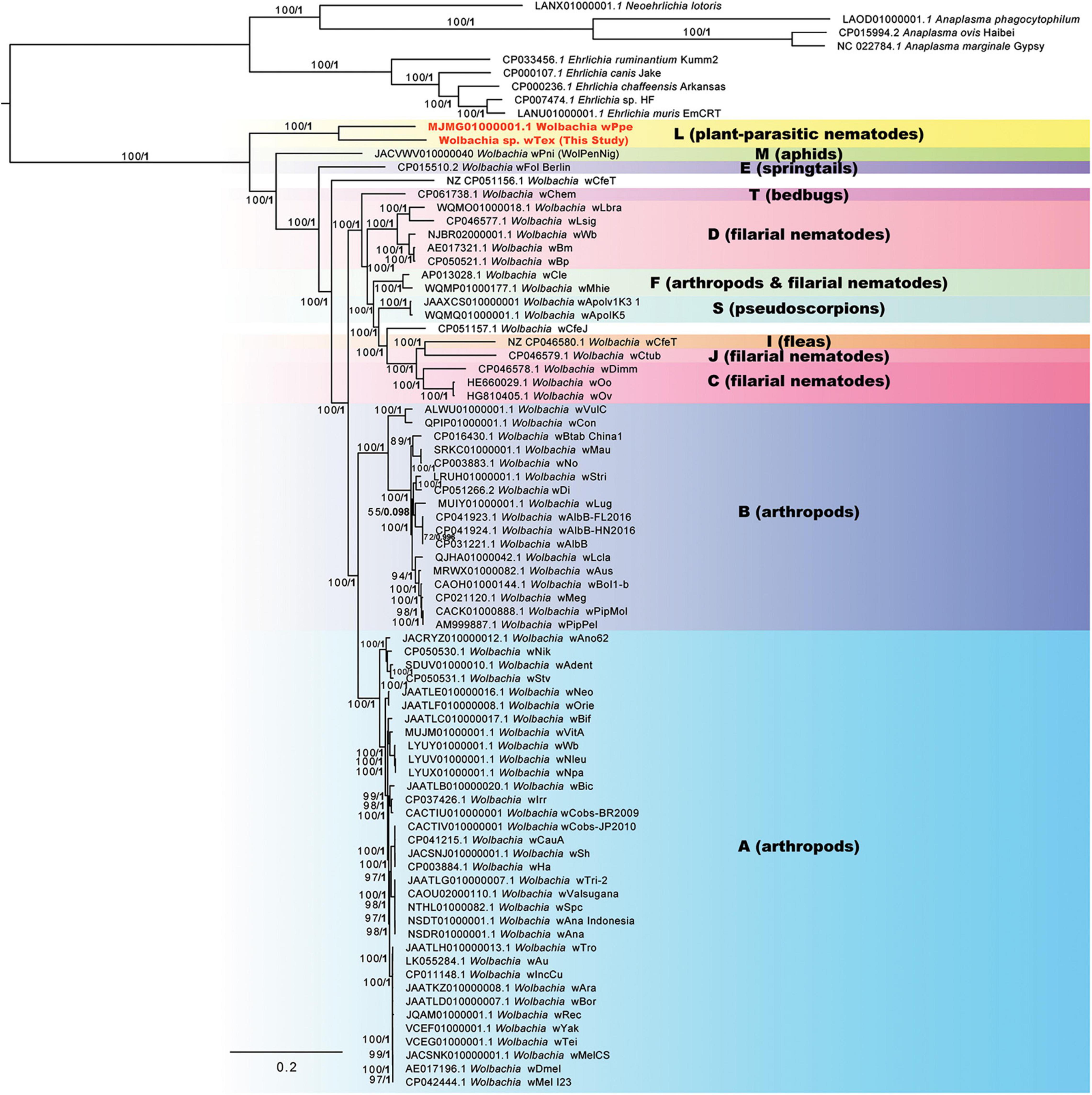
Figure 3. Phylogeny of 100 core protein-coding genes for Wolbachia and outgroups based on 29,264 aligned nucleotide positions identified from assemblies using Roary software, with gaps and ambiguous states and 3rd positions of codons removed. Maximum likelihood phylogeny reconstruction was performed in RAxML GTR + Gamma with bootstrap support >50% from 500 replicates shown on branches. Most supported nodes were obtained with high support in Bayesian 50% majority rule in MrBayes with GTR + G with 4 rate categories model. Bayesian posteriors are shown next to bootstrap values, in bold. Sequences obtained in this study are indicated in bold orange font.
Screening of Wolbachia From Global Soil and Rhizosphere Sequence Read Archive Databases
Sequence read archive (SRA) database screening of 3,400 amplicon experiments from soils and rhizospheres produced 81 sequence runs (i.e., SRR/ERR/DRR files) with the highest blastn similarity to the 16S rRNA gene from PPN-type Wolbachia wTex, wPpe, or wRad. Following read-merging, there was 4,535 top sequences read blastn matches to PPN-type Wolbachia among these runs. After removal of identical sequence reads and sequences leading to exceptionally long branches in phylogenies and sequences placed ambiguously between the Ehrlichia/Anaplasma and Wolbachia clades, there were 61 unique sequences unambiguously grouped with the Wolbachia clade. These sequences were from 24 separate SRA runs originating from the United States, France, Germany, Sweden, Switzerland, Japan, India, and Malaysia. Although phylogenetic analyses with these short sequences produced generally low bootstrap support and low posterior probabilities, tree topologies suggest several clades (Figure 4). Group 1 comprised 19 sequences clustered at the root of the Wolbachia tree along with Wolbachia from PPNs with broad geographic origins (France, Germany, Sweden, Switzerland, Japan, Malaysia, and in the United States, from Florida, Michigan, California, Appalachia) from diverse ecosystems. Groups 2 and 3, from Malaysia and India, clustered near the root of the Wolbachia CDF supergroups. Groups 4 through 8, comprising 28 sequences, were all from a beech forest in Germany and sequences clustered mostly with various Wolbachia strains from Collembola. Group 9 was a distinct cluster of 6 sequences from the same beech forest with similarity to Wolbachia from quill mites, while Group 10 formed a cluster of 6 sequences allied with Wolbachia from Curculio sp. (weevils) from Minnesota. Phylogenetic trees for separate sub-regions produced similar results (Supplementary Figures 5–7).

Figure 4. Phylogeny of plant-parasitic nematode-type Wolbachia-like matches from the SRA database hits 1,047 bp aligned positions of the 16S rRNA gene. Maximum likelihood phylogeny reconstruction was performed in RAxML GTR + Gamma. Bootstrap support >50% from 500 replicates is shown on branches along with posterior probabilities from Bayesian 50% majority rule in MrBayes with GTR + G with 4 rate categories model, shown for several SRA nodes. Sequences from the SRA are indicated in bold blue font and sequences from PPN Wolbachia are indicated in orange bold font. Highlights show clades with Wolbachia-like hits.
Predicted Genes and Pathways Present, Absent, or Unique to Plant-Parasitic Nematode-Type Wolbachia
Specific predicted pathways and genes of interest based on known or hypothesized functions were searched for in the wTex assembly. The wTex genome was similar to wPpe and other Wolbachia in having conserved pathways for glycolysis and the tricarboxylic acid cycle and pathways for biosynthesis of nucleotides including the pentose phosphate pathway, and peptidoglycan and fatty acids, but lacking genes for key steps or most steps of other biosynthetic processes including amino acid, vitamin and co-factor, and carbohydrate synthesis, suggesting incomplete pathways. Both PPN-type Wolbachia strains wTex and wPpe had full-length predicted genes for heme synthesis that were also conserved across outgroups and other Wolbachia, including hemA (encoding 5-aminolevulinate synthase EC 2.3.1.37), hemB (encoding delta-aminolevulinic acid dehydratase EC 4.2.1.24), hemC (encoding porphobilinogen deaminase EC 2.5.1.61), hemE (encoding uroporphyrinogen decarboxylase EC 4.1.1.37), ctaB (encoding protoheme IX farnesyltransferase EC 2.5.1.141), hemF (encoding oxygen-dependent coproporphyrinogen-III oxidase), and hemH (encoding ferrochelatase EC 1.3.3.3).
Riboflavin synthesis and transport genes were notably absent in wTex and wPpe, except for ribB (encoding 3,4-dihydroxy-2-butanone 4-phosphate synthase EC 4.1.99.12) and one outstanding gene annotated as ribN (riboflavin transporter) in wPpe that was not present in any Wolbachia or outgroup strains. Similarly, biotin synthesis genes were absent in wTex and wPpe, with no homologs found matching either outgroup Anaplasmataceae-type biotin genes or the frequently horizontally transferred ‘BOOM’ (biotin synthesis operon of obligate intracellular microbes) operon genes, bioA (encoding adenosylmethionine-8-amino-7-oxononanoate aminotransferase EC 2.6.1.62), bioB (encoding biotin synthase EC 2.8.1.6), bioC (encoding malonyl-[acyl-carrier protein] O-methyltransferase EC 2.1.1.197), bioD (encoding ATP-dependent dethiobiotin synthetase EC 2.6.1.62), bioF (encoding 8-amino-7-oxononanoate synthase 2 EC 2.3.1.47), and bioH (encoding pimeloyl-[acyl-carrier protein] methyl ester esterase EC 3.1.1.85). However, both wTex and wPpe assemblies had a predicted bioY gene (encoding a biotin importing transporter protein), and birA (encoding the biotin ligation protein bifunctional ligase/repressor EC 6.3.4.15), and diverged variants of the biotin utilization genes pccB (encoding propionyl-CoA carboxylase beta chain EC 2.1.3.-) and fabD (encoding malonyl CoA-acyl carrier protein transacylase EC 2.3.1.39). The latter two genes had such low sequence similarity to other Wolbachia strains and outgroups that they were not clustered as homologs to similarly annotated copies in Roary.
Additional predicted genes in wTex that were shared between PPN-type Wolbachia wTex and wPpe, but unique to these strains (i.e., not found in other Wolbachia) included a 1,350 nt gene lysC (encoding lysine-sensitive aspartokinase 3 EC 2.7.2.4, involved in the first step of the lysine biosynthesis via diaminopimelate ‘DAP’ pathway (see Figure 5) which also leads to methionine biosynthesis via de novo pathway, and threonine biosynthesis). This gene had the closest blastn and blastx similarity to genes from the alphaproteobacteria Candidatus Midichloria mitochondrii (hereafter Midichloria) (accession NC_015722.1), or distant genera of Vibrionales (Photobacterium, Vibrio), but no homologs to the standard Wolbachia or Anaplasmataceae variants of the lysC gene. Directly adjacent to this predicted lysC gene was a unique 987 nt PPN-type Wolbachia variant of asd2 (encoding aspartate-semialdehyde dehydrogenase 2 EC 1.2.1.11, which catalyzes the second step in lysine biosynthesis via the DAP pathway), with closest blast matches to genes from Midichloria and next to distantly related bacteria Photobacterium, and Vibrio. Although wPpe had both variants of asd2, wTex had only the Midichloria-like variant. These Midichloria-like asd2 and lysC genes were located adjacent to Wolbachia-like carA (encoding carbamoyl-phosphate synthase small chain EC 6.3.5.5, the first step of pyrimidine and arginine synthesis) and dapA (encoding 4-hydroxy-tetrahydrodipicolinate synthase EC 4.3.3.7, catalyzing the third step in the lysine biosynthesis pathway). The asd2 and lysC gene operon was syntenic, with the same gene order and orientation as that of Midichloria, although the latter had a branched-chain amino acid transaminase (BCAT EC 2.6.1.42) in the place of the dapA in PPN-type Wolbachia, whereas the distantly related versions of asd2 and lysC in other Wolbachia strains and outgroup Anaplasmataceae were not located together in tandem (Supplementary Figure 8). Phylogenetic analyses of all asd2 and lysC variants showed that the PPN-type Wolbachia and Midichloria versions of these genes have phylogenetically diverged from all other Anaplasmataceae/Wolbachia variants with high bootstrap support for a sister-clade to Midichloria for both genes (Supplementary Figures 9, 10). Nucleotide identity between Midichloria-like and PPN-type asd2 and lysC genes was ∼63 and 60%, respectively, while identity between wTex and wPpe homologs were ∼79 and 81%, respectively. Given the finding of these lysine biosynthesis pathway genes (Figure 5), and the observation that the missing final gene required for lysine synthesis lysA (encoding diaminopimelate decarboxylase EC 4.1.1.20) was previously predicted as a bacteria-to-eukaryote HGT in whiteflies (Luan et al., 2015) we searched the complete metagenome assemblies comprising mostly nematode contigs using blastn and blastp to see if we could detect lysA in a contig matching nematodes, but no such match was found. However, coverage was low, leaving some uncertainty about missing genes.
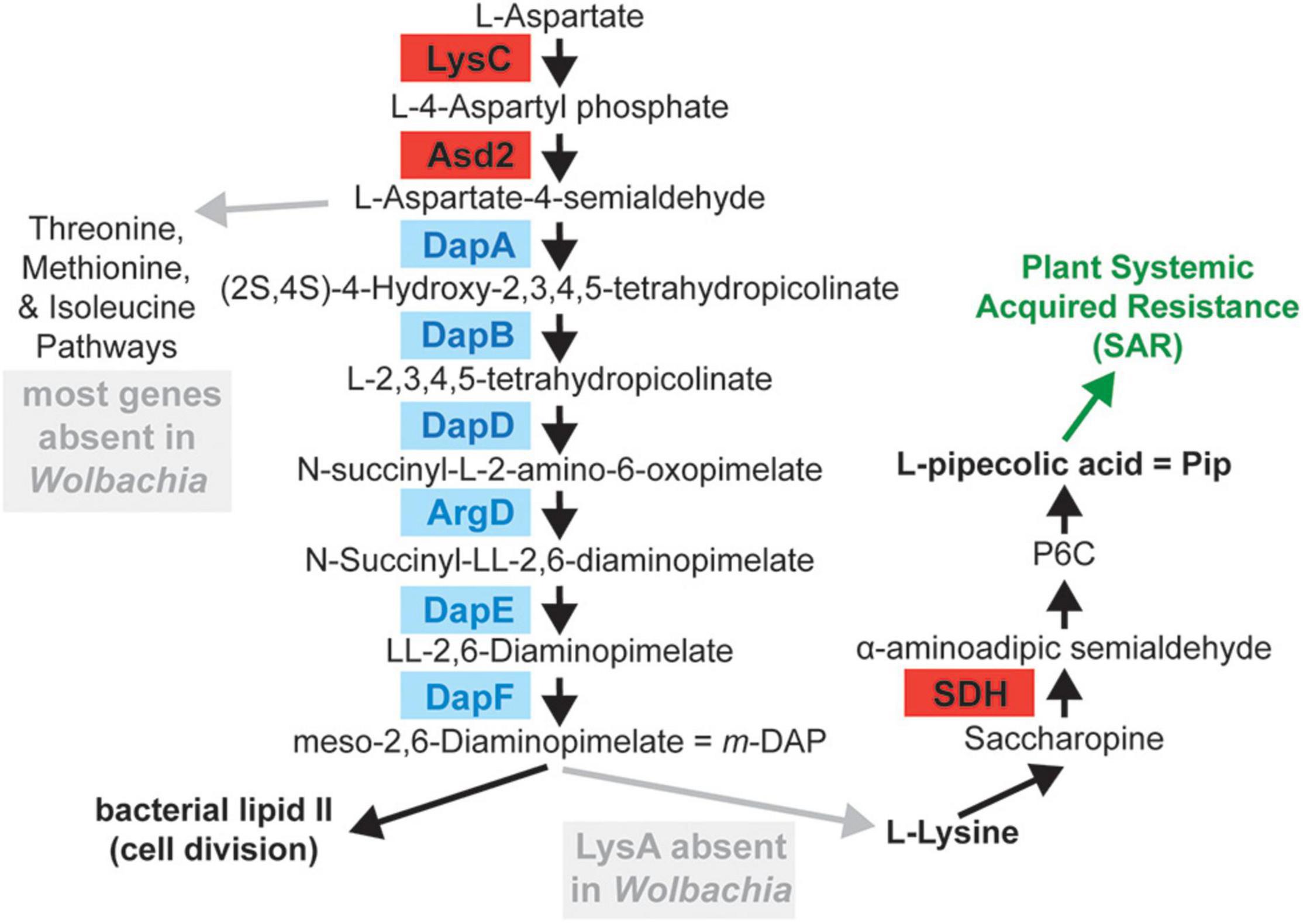
Figure 5. Lysine biosynthesis pathway (diaminopimelate or DAP pathway) and lysine catabolism to pipecolic acid (Pip) showing predicted enzymes that are conserved among plant-parasitic nematode (PPN) Wolbachia, with predicted horizontally transferred genes (HGTs) in red. The genes for LysC and Asd2 genes reflect a putative Midichloria-like HGT, whereas the gene for SDH reflects a putative eukaryote-like HGT to PPN-Wolbachia and also the related early-branching strain wPni. Gray boxes show missing steps likely filled by plant-to-nematode-to-Wolbachia supplementation and gray arrows point to intermediates that could be secreted by PPN-Wolbachia to the host.
Another predicted lysine metabolism gene, shared only among PPN-type Wolbachia (wTex_00187) and wPni, was most similar (67% amino acid identity) to a saccharopine dehydrogenase (SDH) gene (EC 1.5.1.9) in the conifer aphid (Cinara cedri), involved in lysine catabolism (Figure 5). There was also synteny conservation in wTex and wPpe for a 1,308 nt predicted gene (wTex_00248) which had no blastn or blastx matches in the nt/nr databases, which was adjacent to hemA, which catalyzes the first step of heme synthesis. Another unique gene in wTex and wPpe was a 1,494 nt predicted gene tlcA (encoding ADP, ATP carrier protein 1) with its closest match being 70% nucleotide similarity to homologs in Rickettsia. There was a 729 nt predicted gene with partial blastn similarity to a squalene/phytoene synthase/isoprenoid synthase gene (potentially involved in carotenoid synthesis) from the conifer aphid C. cedri and a Wolbachia strain from the gall mite Fragariocoptes setiger. The latter was adjacent to dxr (encoding 1-deoxy-D-xylulose 5-phosphate reductoisomerase EC 1.1.1.267). Two other shared genes predicted in wTex and wPpe were arrayed in synteny: a 663 nt gene annotated as deoC (a gene involved in carbohydrate degradation EC 4.1.2.4) with high blastx similarity to BON (bacterial OsmY and nodulation) domain-containing protein genes in unrelated bacteria such as Flavobacteria, Holosporaceae, Cardinium, and Rickettsia; and an adjacent 246 nt predicted gene with DUF2188 domain and no matches in either blastn or blastx to the nt/nr databases. The first of these two genes had partial length homology (69–80% amino acid identity) to genes from two Wolbachia strains, wCfeT from cat fleas and a strain from the gall mite F. setiger. There were also predicted def (encoding peptide deformylase EC 3.5.1.88) and pterin-4-alpha-carbinolamine dehydratase genes (PCBD1 EC 4.2.1.96) involved in phenylalanine metabolism to tyrosine through tetrahydrobiopterin with distinct homologs in PPN-type Wolbachia that more closely matched non-Wolbachia, including eukaryotes (Culicoides midges) and distantly related bacteria (Francisella), or Rickettsia and an ameba endosymbiont (Candidatus Nucleicultrix amoebiphilia), respectively.
We searched for cytoplasmic incompatibility factor genes, cifA and cifB. From the 92 Wolbachia genomes analyzed in Roary, there were 152 cifA and cifB-like genes identified, yet these shared no homology with any predicted genes in wTex or wPpe or clade C or D Wolbachia. Next, we searched for WO prophage-like or plasmid-associated genes. Strain wTex showed no homology to the more than 2,000 phage or prophage-type genes detected in these 92 Wolbachia genomes analyzed. Nor did wTex have any detected homologs to the 84 plasmid-type genes or in this set of genomes. Other enriched features of PPN-type Wolbachia are described in the section below.
Comparative Genome Repertoires and Gene Ontology Enrichment Analysis
Analysis of gene repertoire overlap between PPN-type Wolbachia strains wTex and wPpe showed 501 predicted shared genes between these strains with an additional 400 and 437 genes only found in wTex and wPpe, respectively. Compared to wPpe, strain wTex was enriched for GO processes DNA-transposition, thiamine biosynthesis, and thiamine diphosphate biosynthesis (Supplementary Table 3), whereas the wPpe strain was enriched for GO processes mismatch repair and one-carbon metabolism (Supplementary Table 4). The thiamine enrichment in wTex arises from three genes, thiE (encoding thiamine-phosphate synthase EC 2.5.1.3), thiM (encoding hydroxyethylthiazole kinase EC 2.7.1.50), and thiD (encoding hydroxymethylpyrimidine/phosphomethylpyrimidine kinase EC 2.7.1.49 EC 2.7.4.7) which occurred as duplication in two scaffolds. Of these genes, only thiM was present in wPpe, whereas the variants of these genes in wTex had close matches only to one Wolbachia strain (wCfeT) and otherwise were most similar to these genes in the spirochete Brachyspira.
Comparison of the shared predicted genes between these two strains (i.e., core genes) compared to the four most closely related strains with complete genome assemblies (namely, wPni from the banana aphid Pentalonia nigronervosa, wFol from the springtail Folsomia candida, wCfeT from the cat flea Ctenocephalides felis, wChem from the bedbug Cimex hemipterus), not counting the 290 universally shared genes among these strains, showed the core PPN-type Wolbachia gene repertoire was most similar to that of wPni, followed by wFol, then wCfeT, and wChem (Figure 6A). This mirrored their phylogenetic places described above, with the four strains having 132, 104, 86, and 66 shared genes, respectively, not counting the 290 universally shared genes between all these strains. GO enrichment for the set of shared core PPN-type Wolbachia genes not shared with these other strains or shared with at most two other strains (Figure 6A) showed enrichment for biological processes diaminopimelate, pseudouridine, and chorismite biosynthesis, gluconeogenesis, and lysine biosynthesis via diaminopimelate, while GO enriched metabolic processes were metallo-aminopeptidase activity, lyase activity, monooxygenase activity, and ATP binding (Supplementary Table 5).
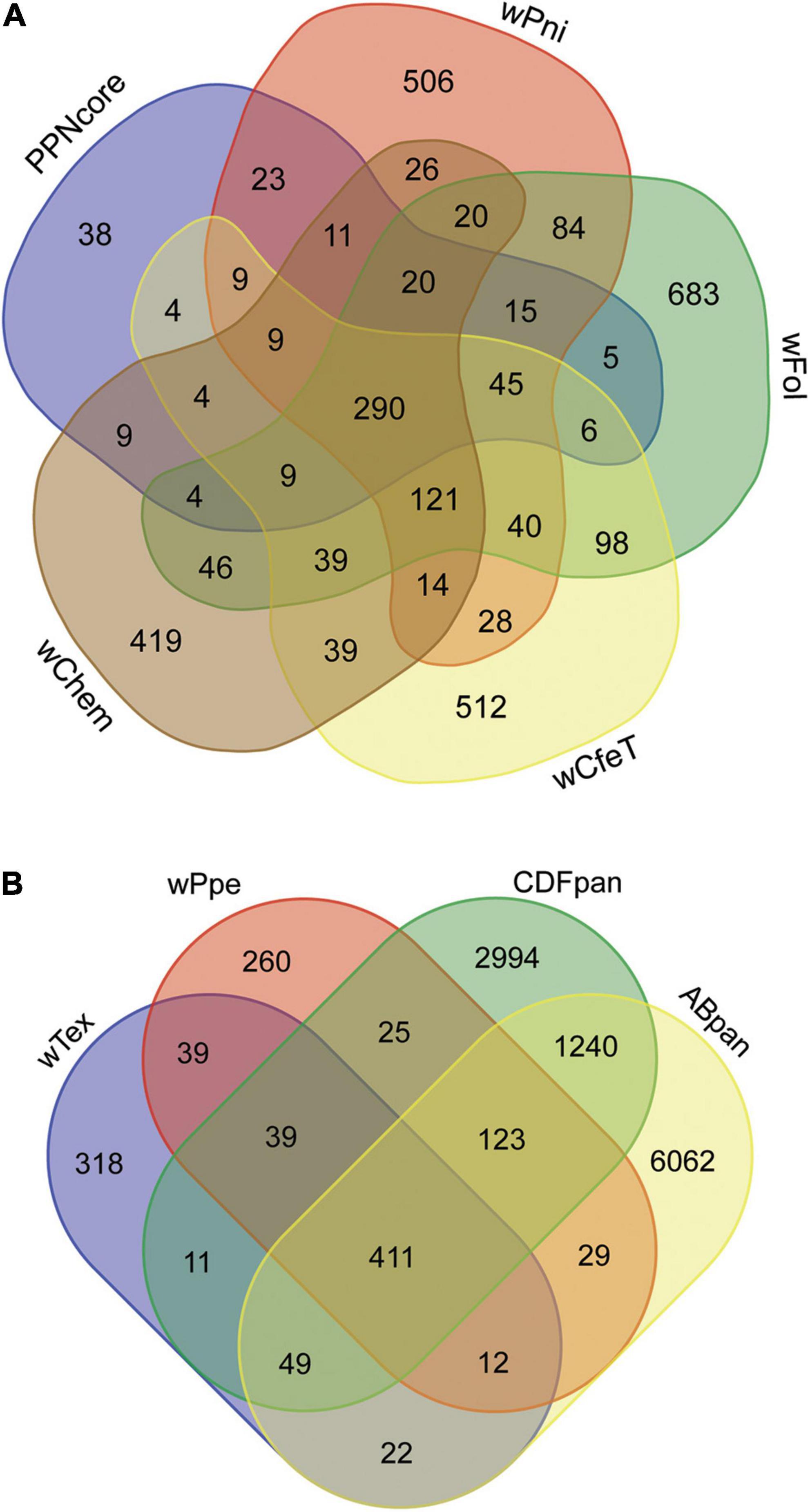
Figure 6. Gene content shared among Wolbachia strains from plant-parasitic nematodes (PPNs) wTex and wPpe, compared with other Wolbachia clades. (A) Depicts core shared genes from PPN Wolbachia compared with pangenomes from other early-branching strains, wPni from the banana aphid Pentalonia nigronervosa, wFol from the springtail Folsomia candida, wCfeT from the cat flea Ctenocephalides felis, wChem from the bedbug Cimex hemipterus. (B) Depicts shared genes among PPN strains wTex and wPpe compared with members of the most widespread supergroups A and B (ABpan), and supergroups with representatives from nematodes, supergroups C, D, and F (CDFpan).
Comparison of genome repertoires of PPN-type Wolbachia strains wTex and wPpe to that of the pangenomes of remaining major supergroup clusters (supergroups C, D, and F denoted “CDFpan” and supergroups A and B denoted “ABpan”) (Figure 6B) showed more core PPN Wolbachia genes shared with CDFpan than with ABpan (683 vs. 649, respectively). Specifically considering genes shared with only CDFpan or ABpan, there was a similar pattern (i.e., 39 for CDFpan vs. 12 for ABpan). The AB pangenome was larger than the CDF pangenome (7,848 vs. 4,892 genes, respectively), therefore alternatively, the differences could be calculated as proportions of each pangenome cluster shared with PPN-type Wolbachia. The latter comparison showed PPN-type Wolbachia shared 0.1396 of their pangenome with CDFpan whereas 0.0817 of their pangenome with ABpan. Conversely, there were more accessory genes (not shared between PPN-type Wolbachia strains) that were shared with ABpan than with CDFpan (22 vs. 11 for wTex and 29 vs. 25 for wPpe), although this difference was not found when controlling for the approximately doubled number of accessory (non-shared) genes in ABpan compared with CDFpan (6,062 vs. 2,994 genes, respectively).
The GO enrichment analyses for PPN-type Wolbachia strains wTex and wPpe compared to other Wolbachia showed several significantly enriched functions (Figure 7 and Supplementary Tables 6–8). GO enrichment was compared for three sets of overlap (Figure 7), first for PPN-type Wolbachia compared to the pangenome of all Wolbachia, including genes shared between pangenomes (Figure 7A and Supplementary Table 6), then for the core shared genes from PPN-type Wolbachia compared to the pangenome of all Wolbachia (Figure 7B and Supplementary Table 7), then for the total pangenomes of PPN-type Wolbachia compared to the pangenome of all Wolbachia (Figure 7C and Supplementary Table 8). Among PPN-type Wolbachia, there was significant GO term enrichment (Figure 7A) for various cellular biosynthetic processes, including several nutrient pathways: protoporphyrinogen IX biosynthesis (part of heme synthesis), thiamine and thiamine diphosphate biosynthesis, lysine biosynthesis via diaminopimelate and diaminopimelate biosynthesis, fatty acid biosynthesis, cellular amino acid biosynthesis. Among GO enrichment for core functions shared by wTex and wPpe (Figure 7B) were many GO terms including nutrient pathways such as protoporphyrinogen IX biosynthesis, lysine biosynthesis via diaminopimelate and diaminopimelate biosynthesis, and fatty acid biosynthesis. For pangenomes of wTex and wPpe excluding shared genes with other Wolbachia (Figure 7C), enriched GO terms included thiamine and thiamine diphosphate biosynthesis, cellular amino acid biosynthesis, and cobalamin biosynthesis.
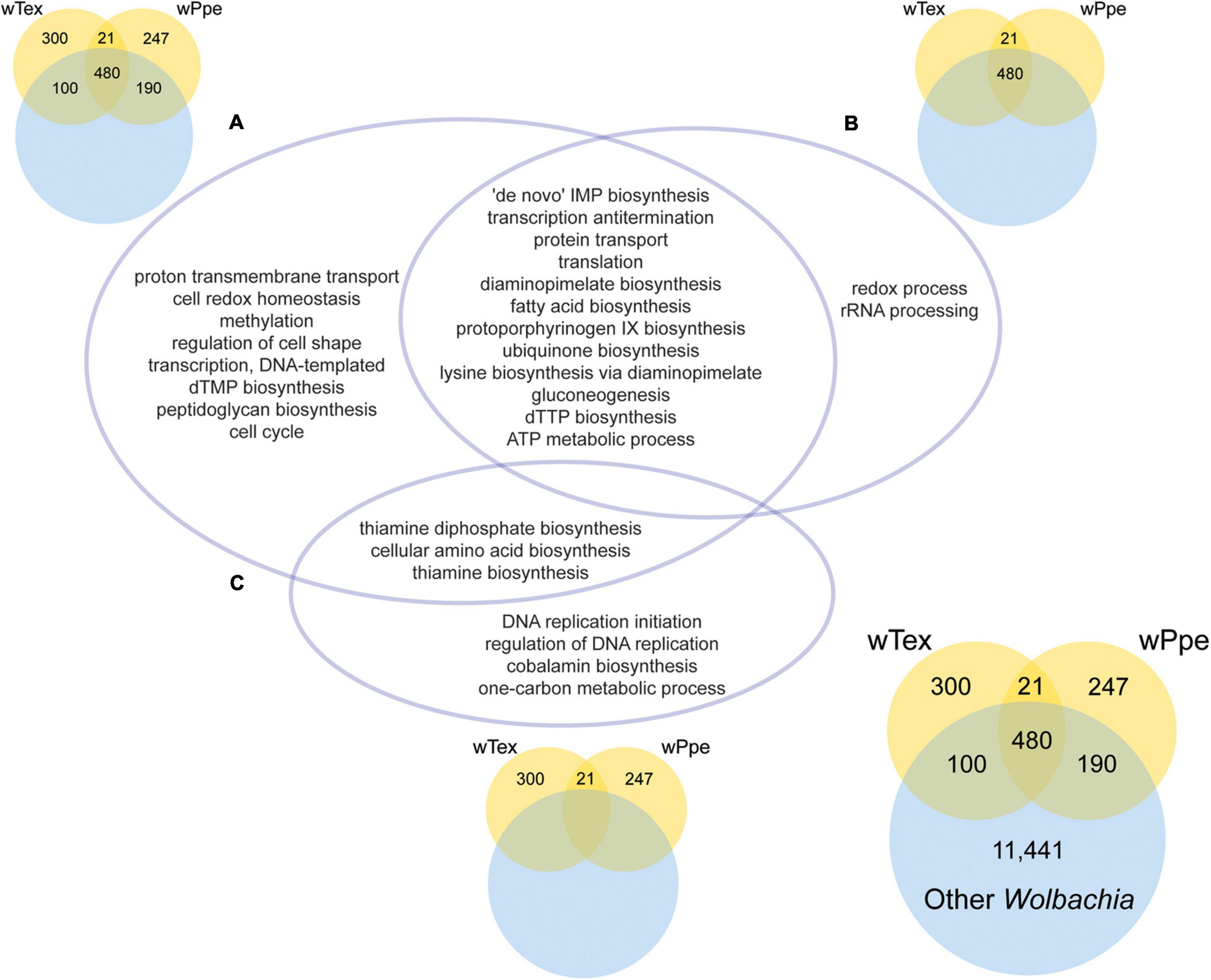
Figure 7. Significantly enriched gene ontology (GO) categories for the pangenomes of plant-parasitic nematode-associated Wolbachia strains (wTex and wPpe) compared to other Wolbachia. The intersections of three GO enrichment tests (A–C) are depicted in the central Venn diagram. (A) Corresponds to the colored Venn on the top left depicting GO enrichment for the PPN pangenome including genes shared with other Wolbachia. (B) Corresponds to the colored Venn on the top right depicting GO enrichment for core shared PPN genes including those shared with other Wolbachia. (C) Corresponds to the colored Venn on the bottom middle depicting GO enrichment for PPN pangenome genes not shared with other Wolbachia. The bottom right Venn shows numbers of genes compared for all groups. Full topGO results are shown in Supplementary Tables 6–8.
Analysis of Changes in Signatures of Selection on Gene Ontology Categories Across Early-Branching Wolbachia Strains
Analysis of dN/dS to investigate signatures of purifying or positive selection within PPN-type Wolbachia (wTex and wPpe) produced a range of dN/dS values from 0.0119336 to 0.334742, with a mean dN/dS of 0.08011301, considering values with Ks < 2. For genes within the dN/dS alignment block that fell within the top or bottom 10% or 25%, topGO enrichment analyses were performed against the pangenome of PPN-type Wolbachia (Supplementary Figure 11 and Supplementary Tables 9–12). Enriched GO categories for the highest values of dN/dS indicative of lower-than-average purifying selection included transcription antitermination, protein folding and transport, and various nutrient metabolism processes including arginine metabolism, N2-acetyl-L-ornithine:2-oxoglutarate 5-aminotransferase activity (a part of arginine metabolism), diaminopimelate biosynthesis (a lysine precursor), lysine biosynthesis, pyridoxal phosphate (vitamin B6) binding (Supplementary Tables 9, 11). Notable GO categories with the lowest dN/dS values (indicative of highest purifying selection) included translation and key metabolic functions associated with an energy (e.g., ATP binding and TCA) and metal ion, iron-sulfur cluster, and heme-binding (Supplementary Tables 10, 12).
The analysis of enriched GO categories for gene sets with highest and lowest dN/dS values associated with transitions between early-branching clades of Wolbachia was analyzed (Figure 8 and Supplementary Tables 13–20). Specifically, universally conserved lowest dN/dS (bottom 10% values) shared among early-branching clades of Wolbachia (strains wTex, wPpe, wPni, wFol, and wCfeT – branch topology and branch lengths extracted in Figure 8 from Figure 3) showed 7 GO categories including DNA topoisomerase type II activity and respirasome. In contrast, conserved lowest dN/dS gene sets showed diverse differences in GO terms unique to each branch. PPN-type Wolbachia strains wTex and wPpe showed unique (not shared) enrichment for heme binding, phosphorelay signal transduction system, ribosome, and zinc ion binding. Uniquely enriched GO terms from the lowest dN/dS gene set in the branch from PPN-type Wolbachia to wPni included 12 terms including protoporphyrinogen IX biosynthetic process. Uniquely low dN/dS gene set GO enrichment in the branch from wPni to wFol included four-way junction helicase and translation elongation factor activity, while the respective enriched terms from wFol to wCfeT were 4 iron, 4 sulfur cluster binding (Figure 8). Various GO categories were uniquely enriched for the highest dN/dS gene sets (top 10%) in these branches, such as for PPN-type Wolbachia, in terms of protein folding and transport, chaperone binding, and 3-dehydroquinate synthase activity (part of aromatic amino acid synthesis via the shikimate pathway) (Figure 8). For the branch from PPN-type Wolbachia to wPni, high dN/dS enrichment included glucosamine-1-phosphate N-acetyltransferase activity (involved in amino sugar metabolism) and many cellular membranes and cell shape/division terms. For the branch from wPni to wFol, high dN/dS enrichment included 4-hydroxy-tetrahydrodipicolinate synthase activity (involved in lysine biosynthesis) and various ribosome and rRNA processing functions, as well as shikimate metabolism (Figure 8). For the branch from wFol to wCfeT, high dN/dS enrichment included 7 terms (Figure 8), including thiamine pyrophosphate binding (involved in thiamine transport).

Figure 8. Enriched gene ontology (GO) categories associated with transitions between early-branching clades of Wolbachia showing GO enrichment for highest and lowest 10% of dN/dS values for genes from pairs of strains wTex, wPpe, wPni, wFol, and wCfeT. Pairwise dN/dS was calculated with KaKs Calculator for overlapping 1,200 bp blocks and GO enrichment was calculated with topGO at p-value < 0.05 and reported for unique GO terms among clades as well as universally conserved (bottom 10%) values. Shading: yellow = unique to plant-parasitic nematode-associated Wolbachia, green = unique to plant-parasitic nematode-associated Wolbachia and wPni, purple = unique to wPni and wFol, blue = unique to wFol and wCfeT. Full topGO results are shown in Supplementary Tables 13–20.
Discussion
While Wolbachia is widespread in insects and filarial nematodes, its presence and importance in rhizosphere hosts are largely unknown. Its distribution and function in plant-parasitic nematodes (PPNs) are of interest evolutionarily, ecologically, and for agriculture as a potential target for developing new biological controls. Here, we screened rhizosphere communities for Wolbachia strains to gain insight into their presence globally. We discovered, assembled, and analyzed a new PPN Wolbachia strain (wTex), comparing functional enrichment and signatures of selection and evaluated genes and genomic patterns that might indicate their role and illuminate their place in the early evolution of this widespread endosymbiont.
Genome features and phylogenetic position of strain wTex confirmed the deep branching place of PPN-type Wolbachia (i.e., supergroup L). Profiling of wTex showed it to be similar to wPpe from the PPN P. penetrans with respect to important features, such as lacking gene matches to cytoplasmic incompatibility (CI) systems (cifA and cifB), WO prophages, plasmid-associated genes, and riboflavin (vitamin B2) genes except for ribB, the horizontally transferred biotin (vitamin B7) ‘BOOM’ operon, yet possessing biotin import and utilization genes (bioY, birA, pccB, and fabD). These features support the hypotheses proposed previously that these widespread, critically important Wolbachia host-impacting traits arose later, after the emergence of PPN Wolbachia via horizontal gene transfers (HGTs) (Nikoh et al., 2014; Moriyama et al., 2015; Brown et al., 2016; Gerth and Bleidorn, 2016; Chen et al., 2020). We found that although supergroup L Wolbachia shared more features (e.g., GC content, assembly length, gene repertoire) with supergroups C, D, and F (comprising mostly obligate mutualists) compared to supergroups A and B, their apparent low prevalence, low titer, suggested by the current study, and possible sex ratio bias phenotype indicated in a previous study (Wasala et al., 2019), suggest they are neither obligate mutualists nor typical CI-inducers. However, our data indicated more unshared than shared genes between PPN Wolbachia strains with a large portion of predicted proteins having unknown functions. Despite this open mystery on these accessory genes, our analyses of the shared genes and annotated portions of these genomes yielded insights into the possible function of these Wolbachia.
Strains wTex and wPpe showed a core gene repertoire most like that of Wolbachia wPni from the plant-feeding host, banana aphid Pentalonia nigronervosa. Furthermore, the closest relatives to PPN-type Wolbachia were from 5 plant-feeding specialist arthropod hosts (genera Pentalonia, Stomaphis, Cinara, Bemisia, and Bryobia), based on available 16S rRNA sequences. Together, these data are consistent with the hypothesis that the earliest Wolbachia symbiosis emerged in plant-feeders, raising the question of whether plant diet specialization drove the early emergence of Wolbachia prior to the later acquisition of genes, such as nutrient supplementation and cytoplasmic incompatibility genes that led to the widespread success of this symbiosis in non-plant feeding hosts. An obstacle to evaluating this hypothesis is that the role of the Wolbachia strains in these plant-specialist hosts remains unclear: previous analysis of wPni suggested it cooperated in nutritional supplementation as a co-mutualist with the aphid primary symbiont Buchnera (De Clerck et al., 2014, 2015), but more recent re-analysis questions this idea (Manzano-Marín, 2020). Nevertheless, despite apparently incomplete essential nutrient biosynthesis pathways in PPN Wolbachia, the possibility remains that intermediate or missing genes that complete these pathways could be present in the nematode hosts, for example, derived from ancient horizontal gene transfers as observed in the Portiera-whitefly symbiosis (Luan et al., 2015; Ankrah and Douglas, 2018). Such HGTs to hosts could also derive from ancient or ancestral bacterial infections (McNulty et al., 2010; Dunning Hotopp, 2011; Brelsfoard et al., 2014; Koutsovoulos et al., 2014; Husnik and McCutcheon, 2018). However, a detailed study of this possibility will require quality PPN genomes and transcriptomes. Nevertheless, the idea that plant diet specialization drove the Wolbachia symbiosis fits with estimated fossil-calibrated Wolbachia divergence (Gerth and Bleidorn, 2016). Approximating ∼216 million years per 2.8% 16S rRNA gene divergence strains wTex, wPpe, and wRad may have diverged from other Wolbachia approximately 314–324 Mya during the high-oxygen mid-Carboniferous period at a time of major nematode diversification, while the wPni-clade Wolbachia may have diverged during the major radiation of insects ∼303 Mya (Rota-Stabelli et al., 2013; Brown et al., 2018).
Connected with this pattern of plant-feeding, our GO enrichment analyses hinted at changes in the plant-limited amino acid lysine in the earliest-branching Wolbachia. Lysine is an essential amino acid that nematodes cannot synthesize de novo but must obtain from their diets, whereas lysine is usually one of the most limiting amino acids in plant diets (Galili, 2002; Galili et al., 2016). Plant roots on which PPNs feed may have especially limited lysine levels due to either lysine catabolism for energy during carbon starvation (Galili et al., 2016), or limited lysine transport to root tissues from the major source in chloroplasts of leaves, or defensive downregulation of lysine synthesis or upregulation of lysine catabolism in roots in response to nematode infection (Pratelli and Pilot, 2014). Thus, one nutrient that PPN-type Wolbachia might be expected to supplement is lysine. Consistent with this idea, we found core genes of PPN-type Wolbachia were significantly enriched for lysine biosynthesis, while the phylogenetic branch from wPni to wFol (representing a transition from Wolbachia in plant-specialists to non-plant specialists) showed decreased purifying selection on lysine biosynthesis (through 4-hydroxy-tetrahydrodipicolinate synthase activity). However, Wolbachia appears to lack the final enzyme required for lysine synthesis, lysA (encoding meso-diaminopimelate decarboxylase), and our analyses found no homolog of lysA in wTex, wPpe, nor any evidence of a lysA-like HGT to their respective hosts. Rather than synthesizing lysine, Wolbachia is thought to use amino acids, including lysine, as a primary energy source (Wu et al., 2004; Foster et al., 2005; Caragata et al., 2016; Ju et al., 2017; Jiménez et al., 2019), thus, the enrichment for lysine pathway genes could reflect increased demand for intermediates (Figure 5), or could implicate an as yet unknown pathway or pathway complementation route to supplement lysine.
Surprisingly, we found PPN-type Wolbachia possessed a conserved Midichloria-like putative HGT for lysine biosynthesis genes. We suggest three alternative hypotheses for this unique dap operon (genes asd2-lysC-dapA) with putative HGTs asd2 and lysC (hereafter, asd2-lysC-HGT). First, the asd2-lysC-HGT could compensate for a prior loss of Anaplasmataceae-like versions lysC found gene to be missing in both PPN-type Wolbachia strains. Curiously, an Anaplasmataceae-like version of asd2 remained in wPpe but was absent in wTex, leaving open the question of the order of loss and gain of the asd2-lysC-HGT and corresponding outgroup genes. Sequencing further strains of early-branching Wolbachia could help address this question. A second possibility is that the genes may serve in some unique manner in PPN-type Wolbachia to augment or affect the late lysine pathway product, meso-diaminopimelate (m-DAP), which is a major constituent of lipid II, which is essential for Wolbachia cell division (Vollmer et al., 2013) (Figure 5). A third possibility is that this conserved asd2-lysC-HGT generates intermediates that are secreted and transferred amongst compartments of the intimate tripartite association of Wolbachia within nematodes within plant roots. Notably, hosts of wPpe and wRad are migratory endoparasites, as are some Helicotylenchus, the possible host of wTex. In this scenario, the secreted intermediates could act as substrates in any aspartate-derived amino acid synthesis, including lysine, in the Wolbachia-PPN-plant tissue niche. Supporting this secretion idea are data suggesting effector and protein secretion from Wolbachia to host cells (Lindsey, 2020) and joint regulation of such secretion systems by host and Wolbachia (Li and Carlow, 2012), as well as evidence that PPN Wolbachia may be localized with the nematode esophageal glands (Brown et al., 2016, 2018), which are specially modified to secrete hundreds of effectors into plant tissues (Vieira and Gleason, 2019). Interestingly, these early pathway enzymes LysC and Asd2 in the aspartate-derived amino acid biosynthesis pathway are highly inducible (Rodionov et al., 2003; Nærdal et al., 2011) and mutational variants in these genes can significantly increase pathway products (Xu et al., 2016). Such controlled intermediate “nutrient factories” from endosymbionts using horizontally transferred genes from other bacteria, including in some cases, missing dap operon genes, lysA, or BCAT (branched-chain amino acid transaminase) have been reported (Luan et al., 2015; Ankrah and Douglas, 2018). Curiously, we found that the asd2-lysC-HGT was adjacent to BCAT in Midichloria but was adjacent to the lysine pathway gene dapA in PPN-type Wolbachia, matching the gene order of distantly related Bacillus spp. and chlamydiae (Rodionov et al., 2003; McCoy et al., 2006). Unexpectedly, this dapA variant was highly similar to the dapA gene in Wolbachia and other Anaplasmataceae, with its gene tree mirroring the species tree, suggesting that this dap operon gene arose earlier in these alphaproteobacteria.
However, the presence of a second lysine-related putative HGT, the eukaryote-like gene sdh for saccharopine dehydrogenase (SDH), in both PPN-type Wolbachia and the early-branching relative from aphids, wPni (Figure 5), suggests a unique host-interaction with lysine catabolism in the early evolution of Wolbachia in plant-feeding hosts. Given the evidence that lysine is often limiting in plants and plant roots (Galili, 2002; Pratelli and Pilot, 2014; Galili et al., 2016) it is curious that the eukaryote-like sdh gene has been conserved. The sdh gene is rare in prokaryotes not living under high osmotic stress (Neshich et al., 2013), so the presence of this gene is unexpected. In Caenorhabditis elegans, saccharopine excess was found to be toxic to mitochondria (Zhou et al., 2019), so it is possible that SDH in these Wolbachia acts to reduce saccharopine toxicity intracellularly. But it would seem unlikely that lysine excess leading to saccharopine excess would be present intracellularly in PPNs if lysine is already limiting in their diets. However, saccharopine excess might arise if there is downregulation of lysine catabolism in roots in response to nematode infection, as has been proposed previously (Pratelli and Pilot, 2014). Alternatively, we suggest the sdh-HGT in these Wolbachia may function to mediate nematode triggering of plant systemic acquired resistance (SAR) through the lysine-to-pipecolic acid (Lys-Pip) system (Návarová et al., 2013; Yang and Ludewig, 2014). Our model for this interaction derives from two sources. First, evidence suggests that plant-parasitic nematodes can induce plant amino acid importers in root cells including amino acid permeases (AAPs AtAAP1-8), lysine/histidine transporters (LHT), and cationic amino acid transporters (CAT AtCAT6) (Hammes et al., 2006; Elashry et al., 2013; Marella et al., 2013), hijacking the existing plant system to acquire limiting aspartate-derived amino acids such as lysine (Yang and Ludewig, 2014). Second, evidence suggests that plants specifically mediate free soluble lysine upon bacterial infection of leaves, first increasing lysine import then massively upregulating lysine catabolism to produce an excess of pipecolic acid (Pip), which then acts as the major metabolic regulator/intensifier of SAR defense (Yang and Ludewig, 2014; Yang et al., 2014) and downregulating lysine synthesis in leaves, reducing this limiting amino acid in roots. Both SAR and decreased lysine will be unfavorable for PPNs and plant pests, thus dysregulating the host-plant Lys-Pip system via secreted SDH from this eukaryote-derived sdh-HGT could be favorable. Further experiments will be needed to assess this hypothesis directly.
Wolbachia genomes from PPNs were also enriched for several other nutrient biosynthesis GO terms including heme and protoporphyrinogen IX (PPG) (part of the heme synthesis pathway). Consistent with gene repertoire enrichment on heme-related pathways, selection analysis examining dN/dS indicated high purifying selection on PPG biosynthesis in PPN-type Wolbachia compared to other Wolbachia. Furthermore, selection analyses also suggested heme-binding was under strong purifying selection in pairwise comparisons between strains wTex and wPpe. Together, these findings are consistent with the ‘iron hypothesis’ for Wolbachia which posits that heme biosynthesis and iron homeostasis may be central to the maintenance of Wolbachia (Foster et al., 2005; Wu et al., 2009; Darby et al., 2012; Gill et al., 2014; Luck et al., 2014; Brown et al., 2016). Nematodes are exceptional among animals in having lost the ability to synthesize heme early in their evolution as bacterivores (Rao et al., 2005; Slatko et al., 2010; Elsworth et al., 2011; Kořený et al., 2021). Most nematodes, as bacterivores, can extract ample heme from their diets. However, descendants of the early bacterivore nematodes such as PPNs and filarial nematodes that evolved to specialize in non-bacterial diets will have had access to limited heme. These PPNs and filarial nematodes are, then, perhaps not surprisingly the only groups to host Wolbachia symbionts. Consistent with the struggle to regain heme in heme-depleted diets, many nematodes have gained a functional HGT of an ancient alphaproteobacterial ferrochelatase gene, the last step in heme synthesis (Wu et al., 2013). Considering our findings, which suggest enhanced essentiality and conservation of heme/PPG in PPN-type Wolbachia, we suggest these Wolbachia heme pathways may have been pivotal in the transition of nematodes to the plant-parasitic lifestyle and may explain the apparent persistence of Wolbachia in certain nematode clades, but not widely across others. It is not clear why genes for PPG biosynthesis would be enriched in PPN-type Wolbachia compared to other groups, including filarial nematodes. However, one possibility is that these PPN Wolbachia strains may generate excess protoporphyrin as a toxin. The observation that Wolbachia wPpe is localized adjacent to the esophageal glands (Brown et al., 2016) could indicate a role for these Wolbachia in producing protoporphyrin destined for nematode secretions during migratory endoparasitic feeding, which may trigger programmed cell death in plant roots by disrupting mitochondrial membranes (Kořený et al., 2021), facilitating nematode feeding.
Wolbachia genomes from PPNs were also enriched for thiamine (vitamin B1) and fatty acid biosynthesis compared to genomes from other Wolbachia. Strain wTex was further enriched for thiamine and thiamine diphosphate biosynthesis genes compared to strain wPpe. This enrichment derived from a multi-gene thiamine synthesis operon (thiE-thiM-thiD) that was only shared with the cat flea Wolbachia wCfeT, but no other Wolbachia, representing a likely HGT from spirochetes. This additional operon suggests some additional thiamine needs in wTex. While thiamine synthesis genes (iscS/adk) are universal in Wolbachia, others, like tenA occur only in a few strains (Lefoulon et al., 2020), whereas Wolbachia in blood-feeding hosts may have acquired genes for thiamine salvage (Nikoh et al., 2014). Thiamine biosynthesis enrichment in PPN-type Wolbachia may derive from a need to supplement this limited essential vitamin, which, like the amino acid lysine, is largely restricted to chloroplast-dense leaves and may be depleted in roots (Martinis et al., 2016). Interestingly, we found in the transition from Wolbachia from wFol from springtails to wCfeT from cat fleas, there appeared to be higher than expected dN/dS on thiamine pyrophosphate binding, which is involved in thiamine transport, suggesting a possible change in thiamine needs in these basal branches of Wolbachia. Conversely, interpreting the enriched fatty acid biosynthesis in PPN-type Wolbachia is more difficult; there are no known fatty acids that are essential (not synthesized) by nematodes (Zečić et al., 2019). However, fatty acids are likely absorbed by nematodes and their uptake could be variable among parasitic nematodes (Mondal et al., 2016) depending on the availability in roots, or potentially through supply by Wolbachia.
To investigate possible functions linked to Wolbachia’s success in early-branching clades, we analyzed other patterns in purifying selection indicated by measures of dN/dS. As might be expected, we found universally high purifying selection on housekeeping activities such as ribonucleotide binding, DNA topoisomerase type II, and DNA topological change, but early-branching Wolbachia also showed enhanced purifying selection for energy and respiration-related activities including ATP binding, NADH dehydrogenase activity, quinone binding, and respirasome activity. These conserved respiration functions may relate to host mitochondria-Wolbachia interaction homeostasis, which has been shown as critical to maintenance in host cells – and disruption of oxidative phosphorylation leading to host cell death in alternate hosts (Uribe-Alvarez et al., 2019). Among wTex and wPpe, there appeared to be the highest purifying selection on metabolic functions associated with energy, and metal ion, iron-sulfur cluster, heme binding, which again point to oxygenic-mitochondrial and heme synthesis processes and mitochondria-Wolbachia interaction and homeostasis as key functions specifically conserved in PPN Wolbachia. Conversely, results showed lower purifying selection – or potentially, directional selection – within wTex and wPpe for various nutrient metabolism processes including arginine metabolism, lysine biosynthesis, and binding of vitamin B6, heme, and zinc ions, as well as protein folding and transport, chaperone binding, and aromatic amino acid synthesis, suggesting these processes, which likely influence host-Wolbachia interactions, are uniquely important in supergroup L Wolbachia. Arginine biosynthesis, however, was under higher purifying selection in the subsequent branch between PPN-type Wolbachia and strain wPni, as processed including protein transport by the Tat complex and proteolysis, suggesting these are core, conserved functions in these early-branching Wolbachia clades. Conversely, higher dN/dS pathways between supergroup L and wPni suggested evolutionary changes in amino ugar metabolism and many cell shape/division functions in these early Wolbachia host transitions.
While Wolbachia-like gene transfers to their eukaryote hosts have been reported in numerous studies (McNulty et al., 2010; Dunning Hotopp, 2011; Brelsfoard et al., 2014; Koutsovoulos et al., 2014; Husnik and McCutcheon, 2018), there has been limited study of HGTs to Wolbachia from other microbes. Our results here suggest some of these hypothesized HGTs may be important. For example, in addition to the putative HGTs for lysine synthesis (asd2-lysC), sdh, and thiamine synthesis (thiE-thiM-thiD), which dN/dS patterns suggest are under enhanced purifying selection, we found other conserved putative HGTs of interest. For example, both PPN Wolbachia shared a predicted large HGT from Rickettsia for the gene tlcA, which is critically important for parasitism-related ATP import or exchange in Rickettsia (Audia and Winkler, 2006; Renvoisé et al., 2011). This tlcA-HGT is curiously absent in other Wolbachia and Anaplasmataceae. There was also a squalene/phytoene synthase gene of putative HGT origin, with the closest match to carotenoid synthesis-associated genes from the conifer aphid C. cedri (Nováková and Moran, 2012), adjacent to dxr, which is an essential gene in the MEP pathway of isoprenoid synthesis and occurs across Wolbachia. It is unclear how these genes, or others such as the eukaryote-like PCBD1-HGT which may act in phenylalanine metabolism to tyrosine, function in PPN Wolbachia. In the future, we suggest the systematic study of such genes would be warranted. To improve such studies, efforts should focus on overcoming genome completeness for MAG datasets. While our wTex genome may be incomplete, due to low coverage and variance among strains from pooled hosts, we expect most of our findings and conclusions discussed here are conservative in that the essential and shared genes were likely detected through our assembly method that centered on extracting contigs with blastn matches to wPpe, Wolbachia, and outgroups. In contrast, we predict that the genes most likely to be missed in our wTex assembly by these methods would be those that are most diverged and regions with novel HGTs that do not map to other Wolbachia. Future work should focus on improving coverage and read length to overcome these issues.
Besides function, our study sought to investigate the distribution of PPN Wolbachia. Wolbachia wTex was found with shallow read depth within these nematode community assemblies, however, relative to the predicted nematode host which had COI gene coverage of ∼2–8× per sample, it had similar coverage per sample (∼0.7–8×) which suggests a high titer in its hosts, based on an estimation of ∼300 mitochondria per host cell. Furthermore, although Wolbachia-like sequence reads were obtained in 10/16 of our sampled sites, only one site had sequences matching PPN-type Wolbachia, despite the presence of hundreds of different nematodes in these samples, based on COI profiling. However, for the Wolbachia-positive site, which was a tropical fruit farm in southern Texas, wTex was in nematode communities from several plants, suggesting its host is not specific to plant species. The possible host of wTex may be the spiral nematode, Helicotylenchus sp., based on abundance correlation analysis. Helicotylenchus spp. are sometimes ectoparasitic, but some species are migratory and burrowing (e.g., the banana spiral nematode Helicotylenchus multicinctus), a lifestyle resembling hosts of other PPN Wolbachia, P. penetrans, and R. similis. This scenario puts a spotlight on future studies of how Wolbachia may play a role-specific to this migratory endoparasitic lifestyle.
Screening of SRA data revealed potential PPN Wolbachia in global soils and rhizospheres. We found the prevalence at about 0.42% of samples, but with few reads per run, suggesting these Wolbachia occur at low titers, at least at the bulk community level. However, these data likely under represent PPN Wolbachia diversity, prevalence, and titer because nematode distribution is patchy, with typical soil samples (∼0.25 g) not capturing much diversity (Donn et al., 2008) and common soil DNA isolation practices often failing to break nematode cuticles (Waeyenberge et al., 2019; Sloan et al., 2021). Universal primer-based amplicon sequencing for PPN Wolbachia is an improvement over PCR methods because most Wolbachia-specific 16S PCR primers have significant mismatches to these early-branching strains (e.g., primers Wolb-SpecF and SpecR and Wol-F-1992 and Wol-R-1992; and the primers Wol_16S_F and Wol_281B_F which match wPpe have mismatches to wTex). However, the major limitation of amplicon data mining is the short length of amplicon sequencing reads which limits the information gained, compared to more costly WGS methods.
Conclusion
This study expands our understanding of early-branching Wolbachia, pointing to unique genes and pathways that give insights into the functions of these elusive PPN Wolbachia strains. Examples include conserved putative HGTs for lysine, thiamine, and heme/protoporphyrinogen IX biosynthesis and genes that may interact with plant immunity, and other enriched pathways with distinct signatures of selection. These enrichment analyses add to the tool set that should be useful for future studies on new Wolbachia. Our community WGS and SRA screens illuminate the broad global and phylogenetic distribution of PPN-type Wolbachia. One major focus for future study of these early-branching Wolbachia will be to investigate the ∼40% of predicted genes that were recovered with no match to genes with known function. Other key questions that require further work are the fitness effects of these Wolbachia on their hosts, which will require improved lab culturing of the nematodes.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, JAIXMJ000000000 and https://www.ncbi.nlm.nih.gov/, PRJNA687334.
Author Contributions
NW and SA assisted with nematode isolation, PCR, and genomic library preparation. AB led the design of experiments, developed bioinformatics code, and pipelines, and drafted the manuscript. All co-authors assisted with the revision of the final manuscript.
Funding
Graduate funding support to SA was through TTUASM, the TTU Graduate Student Research Award, and the Helen DeVitt Jones Graduate Fellowship. Funding to AB was through startup funding from the Department of Biological Sciences at Texas Tech and the National Science Foundation (Award 2047684) and the United States Department of Agriculture NIFA (Award 20216701335757).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank L. T. E. O. Rogers for assistance with field collection, and Robin Ronson and William Barela for assistance with SRA data mining code.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.867392/full#supplementary-material
Footnotes
- ^ https://github.com/tseemann/barrnap
- ^ https://github.com/rambaut/figtree/releases
- ^ https://github.com/lyijin/topGO_pipeline/
References
Adam, M., Westphal, A., Hallmann, J., and Heuer, H. (2014). Specific microbial attachment to root knot nematodes in suppressive soil. Appl. Environ. Microbiol. 80, 2679–2686. doi: 10.1128/AEM.03905-13
Alexa, A., and Rahnenfuhrer, J. (2020). Topgo: Enrichment Analysis for Gene Ontology. R package version 2.40.0.
Ankrah, N. Y. D., and Douglas, A. E. (2018). Nutrient factories: metabolic function of beneficial microorganisms associated with insects. Environ. Microbiol. 20, 2002–2011. doi: 10.1111/1462-2920.14097
Audia, J. P., and Winkler, H. H. (2006). Study of the five rickettsia prowazekii proteins annotated as ATP/ADP translocases (Tlc): Only Tlc1 transports ATP/ADP, while Tlc4 and Tlc5 transport other ribonucleotides. J. Bacteriol. 188, 6261–6268. doi: 10.1128/JB.00371-06
Augustinos, A. A., Santos-Garcia, D., Dionyssopoulou, E., Moreira, M., Papapanagiotou, A., Scarvelakis, M., et al. (2011). Detection and characterization of Wolbachia infections in natural populations of aphids: is the hidden diversity fully unraveled? PLoS One 6:e28695. doi: 10.1371/journal.pone.0028695
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. doi: https://doi.org/10.1111/j.2517-6161.1995.tb02031.x
Bennett, G. M., Heath-Heckman, E., and Sogin, E. M. (2021). Finding needles in haystacks and inferring their function: challenges and successes in beneficial symbiosis research. Msystems 6, 20–23. doi: 10.1128/msystems.00243-21
Bennett, G. M., and Moran, N. A. (2013). Small, smaller, smallest: the origins and evolution of ancient dual symbioses in a phloem-feeding insect. Genome Biol. Evol. 5, 1675–1688. doi: 10.1093/gbe/evt118
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bongers, T., and Ferris, H. (1999). Nematode community structure as a bioindicator in environmental monitoring. Trends Ecol. Evol. 14, 224–228. doi: 10.1016/S0169-5347(98)01583-3
Bordenstein, S. R., Fitch, D. H. A., and Werren, J. H. (2003). Absence of Wolbachia in nonfilariid nematodes. J. Nematol. 35, 266–270.
Brankovics, B., Zhang, H., van Diepeningen, A. D., van der Lee, T. A. J., Waalwijk, C., and de Hoog, G. S. (2016). GRAbB: Selective assembly of genomic regions, a new niche for genomic research. PLoS Comput. Biol. 12:e1004753. doi: 10.1371/journal.pcbi.1004753
Brelsfoard, C., Tsiamis, G., Falchetto, M., Gomulski, L. M., Telleria, E., Alam, U., et al. (2014). Presence of Extensive Wolbachia symbiont insertions discovered in the genome of its host Glossina morsitans morsitans. PLoS Negl. Trop. Dis 8:e2728. doi: 10.1371/journal.pntd.0002728
Brown, A. M. V., Howe, D. K., Wasala, S. K., Peetz, A. B., Zasada, I. A., and Denver, D. R. (2015). Comparative genomics of a plant-parasitic nematode endosymbiont suggest a role in nutritional symbiosis. Genome Biol. Evol. 7, 2727–2746. doi: 10.1093/gbe/evv176
Brown, A. M. V., Wasala, S. K., Howe, D. K., Peetz, A. B., Zasada, I. A., and Denver, D. R. (2016). Genomic evidence for plant-parasitic nematodes as the earliest Wolbachia hosts. Sci. Rep. 6:34955. doi: 10.1038/srep34955
Brown, A. M. V., Wasala, S. K., Howe, D. K., Peetz, A. B., Zasada, I. A., and Denver, D. R. (2018). Comparative genomics of Wolbachia–Cardinium dual endosymbiosis in a plant-parasitic nematode. Front. Microbiol. 9:2482. doi: 10.3389/fmicb.2018.02482
Brown, A. M. V. (2018). Endosymbionts of plant-parasitic nematodes. Annu. Rev. Phytopathol. 56, 225–242.
Bushnell, B. (2014). BBMap: A fast, Accurate, Splice-Aware Aligner. No. LBNL-7065E. Berkeley, CA: Lawrence Berkeley National Laboratory.(LBNL).
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Caragata, E. P., Dutra, H. L. C., and Moreira, L. A. (2016). Exploiting intimate relationships: controlling mosquito-transmitted disease with Wolbachia. Trends Parasitol. 32, 207–218. doi: 10.1016/j.pt.2015.10.011
Carey, J. (2018). News feature: the race to extinguish insect pests by enlisting their own kind. Proc. Natl. Acad. Sci. 115, 7839–7843. doi: 10.1073/pnas.1811000115
Chen, H., Zhang, M., and Hochstrasser, M. (2020). The biochemistry of cytoplasmic incompatibility caused by endosymbiotic bacteria. Genes (Basel) 11, 1–22. doi: 10.3390/genes11080852
Cheng, X.-Y. Y., Tian, X.-L. L., Wang, Y.-S. S., Lin, R.-M. M., Mao, Z.-C. C., Chen, N., et al. (2013). Metagenomic analysis of the pinewood nematode microbiome reveals a symbiotic relationship critical for xenobiotics degradation. Sci. Rep. 3, 1–10. doi: 10.1038/srep01869
Cummins, C. A., and McInerney, J. O. (2011). A method for inferring the rate of evolution of homologous characters that can potentially improve phylogenetic inference, resolve deep divergence and correct systematic biases. Syst. Biol. 60, 833–844. doi: 10.1093/sysbio/syr064
Darby, A. C., Armstrong, S. D., Bah, G. S., Kaur, G., Hughes, M. A., Kay, S. M., et al. (2012). Analysis of gene expression from the Wolbachia genome of a filarial nematode supports both metabolic and defensive roles within the symbiosis. Genome Res. 22, 2467–2477. doi: 10.1101/gr.138420.112
Darling, A. E., Mau, B., and Perna, N. T. (2010). Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
De Clerck, C., Fujiwara, A., Joncour, P., Léonard, S., Félix, M. L., Francis, F., et al. (2015). A metagenomic approach from aphid’s hemolymph sheds light on the potential roles of co-existing endosymbionts. Microbiome 3:63. doi: 10.1186/s40168-015-0130-5
De Clerck, C., Tsuchida, T., Massart, S., Lepoivre, P., Francis, F., and Jijakli, M. H. (2014). Combination of genomic and proteomic approaches to characterize the symbiotic population of the banana aphid (hemiptera: aphididae). Environ. Entomol. 43, 29–36. doi: 10.1603/EN13107
Denver, D. R., Brown, A. M. V., Howe, D. K., Peetz, A. B., and Zasada, I. A. (2016). Genome skimming: a rapid approach to gaining diverse biological insights into multicellular pathogens. PLoS Pathog. 12:e1005713. doi: 10.1371/journal.ppat.1005713
Donn, S., Griffiths, B. S., Neilson, R., and Daniell, T. J. (2008). DNA extraction from soil nematodes for multi-sample community studies. Appl. Soil Ecol. 38, 20–26. doi: 10.1016/j.apsoil.2007.08.006
Douglas, A. E. (2015). Multiorganismal Insects: diversity and function of resident microorganisms. Annu. Rev. Entomol. 60, 17–34. doi: 10.1146/annurev-ento-010814-020822
Dunning Hotopp, J. C. (2011). Horizontal gene transfer between bacteria and animals. Trends Genet. 27, 157–163. doi: 10.1016/j.tig.2011.01.005.Horizontal
Elashry, A., Okumoto, S., Siddique, S., Koch, W., Kreil, D. P., and Bohlmann, H. (2013). The AAP gene family for amino acid permeases contributes todevelopment of the cyst nematode Heterodera schachtii in roots of Arabidopsis. Plant Physiol. Biochem. 70, 379–386. doi: 10.1016/j.plaphy.2013.05.016
Elhady, A., Giné, A., Topalovic, O., Jacquiod, S., Sørensen, S. J., Sorribas, F. J., et al. (2017). Microbiomes associated with infective stages of root-knot and lesion nematodes in soil. PLoS One 12:e0177145. doi: 10.1371/journal.pone.0177145
Elsworth, B., Wasmuth, J., and Blaxter, M. (2011). NEMBASE4: The nematode transcriptome resource. Int. J. Parasitol. 41, 881–894. doi: 10.1016/j.ijpara.2011.03.009
Ferris, H., and Tuomisto, H. (2015). Unearthing the role of biological diversity in soil health. Soil Biol. Biochem. 85, 101–109. doi: 10.1016/j.soilbio.2015.02.037
Foster, J., Ganatra, M., Kamal, I., Ware, J., Makarova, K., Ivanova, N., et al. (2005). The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 3:e121. doi: 10.1371/journal.pbio.0030121
Galili, G. (2002). New insights into the regulation and functional significance of lysine metabolism in plants. Annu. Rev. Plant Biol. 53, 27–43. doi: 10.1146/annurev.arplant.53.091401.110929
Galili, G., Amir, R., and Fernie, A. R. (2016). The regulation of essential amino acid synthesis and accumulation in plants. Annu. Rev. Plant Biol. 67, 153–178. doi: 10.1146/annurev-arplant-043015-112213
Gerth, M., and Bleidorn, C. (2016). Comparative genomics provides a timeframe for Wolbachia evolution and exposes a recent biotin synthesis operon transfer. Nat. Microbiol. 2:16241. doi: 10.1038/nmicrobiol.2016.241
Gill, A. C., Darby, A. C., and Makepeace, B. L. (2014). Iron necessity: the secret of Wolbachia’s Success? PLoS Negl. Trop. Dis. 8:e3224. doi: 10.1371/journal.pntd.0003224
Gressel, J. (2018). Microbiome facilitated pest resistance: potential problems and uses. Pest. Manag. Sci. 74, 511–515. doi: 10.1002/ps.4777
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haegeman, A., Vanholme, B., Jacob, J., Vandekerckhove, T. T. M., Claeys, M., Borgonie, G., et al. (2009). An endosymbiotic bacterium in a plant-parasitic nematode: member of a new Wolbachia supergroup. Int. J. Parasitol. 39, 1045–1054. doi: 10.1016/j.ijpara.2009.01.006
Hammes, U. Z., Nielsen, E., Honaas, L. A., Taylor, C. G., and Schachtman, D. P. (2006). AtCAT6, a sink-tissue-localized transporter for essential amino acids in Arabidopsis. Plant J. 48, 414–426. doi: 10.1111/j.1365-313X.2006.02880.x
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Husnik, F., and McCutcheon, J. P. (2018). Functional horizontal gene transfer from bacteria to eukaryotes. Nat. Rev. Microbiol. 16, 67–79. doi: 10.1038/nrmicro.2017.137
Jenkins, W. R. (1964). A rapid centrifugal-flotation technique for separating nematodes from soil. Plant Dis. Report. 48, 692.
Jiménez, N. E., Gerdtzen, Z. P., Olivera-Nappa, Á, Salgado, J. C., and Conca, C. (2019). A systems biology approach for studying Wolbachia metabolism reveals points of interaction with its host in the context of arboviral infection. PLoS Negl. Trop. Dis 13:e0007678. doi: 10.1371/journal.pntd.0007678
Ju, J. F., Hoffmann, A. A., Zhang, Y. K., Duan, X. Z., Guo, Y., Gong, J. T., et al. (2017). Wolbachia-induced loss of male fertility is likely related to branch chain amino acid biosynthesis and iLvE in Laodelphax striatellus. Insect Biochem. Mol. Biol. 85, 11–20. doi: 10.1016/j.ibmb.2017.04.002
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kořený, L., Oborník, M., Horáková, E., Waller, R. F., and Lukeš, J. (2021). The convoluted history of haem biosynthesis. Biol. Rev. 44, 141–162. doi: 10.1111/brv.12794
Koutsovoulos, G., Makepeace, B., Tanya, V. N., and Blaxter, M. M. (2014). Palaeosymbiosis revealed by genomic fossils of Wolbachia in a strongyloidean nematode. PLoS Genet. 10:e1004397. doi: 10.1371/journal.pgen.1004397
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Lefoulon, E., Clark, T., Guerrero, R., Cañizales, I., Cardenas-Callirgos, J. M., Junker, K., et al. (2020). Diminutive, degraded but dissimilar: Wolbachia genomes from filarial nematodes do not conform to a single paradigm. Microb. Genomics 6, 1–21. doi: 10.1099/mgen.0.000487
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and samtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, Z., and Carlow, C. K. S. (2012). Characterization of transcription factors that regulate the type IV secretion system and riboflavin biosynthesis in Wolbachia of Brugia malayi. PLoS One 7:e51597. doi: 10.1371/journal.pone.0051597
Lindsey, A. R. I. (2020). Sensing, signaling, and secretion: a review and analysis of systems for regulating host interaction in wolbachia. Genes (Basel) 11, 1–21. doi: 10.3390/genes11070813
Löytynoja, A. (2014). Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 1079, 155–170. doi: 10.1007/978-1-62703-646-7_10
Luan, J.-B. B., Chen, W., Hasegawa, D. K., Simmons, A. M., Wintermantel, W. M., Ling, K.-S. S., et al. (2015). Metabolic coevolution in the bacterial symbiosis of whiteflies and related plant sap-feeding insects. Genome Biol. Evol. 7, 2635–2647. doi: 10.1093/gbe/evv170
Luck, A. N., Evans, C. C., Riggs, M. D., Foster, J. M., Moorhead, A. R., Slatko, B. E., et al. (2014). Concurrent transcriptional profiling of Dirofilaria immitis and its Wolbachia endosymbiont throughout the nematode life cycle reveals coordinated gene expression. BMC Genomics 15:1041. doi: 10.1186/1471-2164-15-1041
Manzano-Marín, A. (2020). No evidence for Wolbachia as a nutritional co-obligate endosymbiont in the aphid Pentalonia nigronervosa. Microbiome 8, 1–5. doi: 10.1186/s40168-020-00865-2
Marella, H. H., Nielsen, E., Schachtman, D. P., and Taylor, C. G. (2013). The amino acid permeases AAP3 and AAP6 are involved in root-knot nematode parasitism of arabidopsis. Mol. Plant-Microbe Interact. 26, 44–54. doi: 10.1094/MPMI-05-12-0123-FI
Martinis, J., Gas-Pascual, E., Szydlowski, N., Crèvecoeur, M., Gisler, A., Bürkle, L., et al. (2016). Long-distance transport of thiamine (vitamin b1) is concomitant with that of polyamines. Plant Physiol. 171, 542–553. doi: 10.1104/pp.16.00009
McCoy, A. J., Adams, N. E., Hudson, A. O., Gilvarg, C., Leustek, T., and Maurelli, A. T. (2006). L,L-diaminopimelate aminotransferase, a trans-kingdom enzyme shared by Chlamydia and plants for synthesis of diaminopimelate/lysine. Proc. Natl. Acad. Sci. U.S.A. 103, 17909–17914. doi: 10.1073/pnas.0608643103
McNulty, S. N., Foster, J. M., Mitreva, M., Hotopp, J. C. D., Martin, J., Fischer, K., et al. (2010). Endosymbiont DNA in endobacteria-free filarial nematodes indicates ancient horizontal genetic transfer. PLoS One 5:e11029. doi: 10.1371/journal.pone.0011029
Mee, P. T., Weeks, A. R., Walker, P. J., Hoffmann, A. A., and Duchemin, J. B. (2015). Detection of low-level Cardinium and Wolbachia infections in Culicoides. Appl. Environ. Microbiol. 81, 6177–6188. doi: 10.1128/AEM.01239-15
Mondal, M., Kundu, J. K., and Misra, K. K. (2016). Variation in lipid and fatty acid uptake among nematode and cestode parasites and their host, domestic fowl: host–parasite interaction. J. Parasit. Dis. 40, 1494–1518. doi: 10.1007/s12639-015-0718-5
Moran, N. A., McCutcheon, J. P., and Nakabachi, A. (2008). Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42, 165–190. doi: 10.1146/annurev.genet.41.110306.130119
Moriyama, M., Nikoh, N., Hosokawa, T., and Fukatsu, T. (2015). Riboflavin provisioning underlies wolbachia’s fitness contribution to its insect host. MBio 6, e1732–e1715. doi: 10.1128/mBio.01732-15
Myers, K. N., Conn, D., and Brown, A. M. V. (2021). Essential amino acid enrichment and positive selection highlight endosymbiont’s role in a global virus-vectoring pest. Msystems 6, 1–22. doi: 10.1128/msystems.01048-20
Nærdal, I., Netzer, R., Ellingsen, T. E., and Brautaset, T. (2011). Analysis and manipulation of aspartate pathway genes for L-lysine overproduction from methanol by Bacillus methanolicus. Appl. Environ. Microbiol. 77, 6020–6026. doi: 10.1128/AEM.05093-11
Návarová, H., Bernsdorff, F., Döring, A. C., and Zeier, J. (2013). Pipecolic acid, an endogenous mediator of defense amplification and priming, is a critical regulator of inducible plant immunity. Plant Cell 24, 5123–5141. doi: 10.1105/tpc.112.103564
Neshich, I. A., Kiyota, E., and Arruda, P. (2013). Genome-wide analysis of lysine catabolism in bacteria reveals new connections with osmotic stress resistance. ISME J. 7, 2400–2410. doi: 10.1038/ismej.2013.123
Nicol, J. M., Turner, S. J., Coyne, D. L., Nijs, L., den Hockland, S., and Tahna Maafi, Z. (2011). in Genomics and Molecular Genetics of Plant-Nematode Interactions, eds J. T. Jones, G. Gheysen, and C. Fenoll Heidelberg (New York, NY: Springer), doi: 10.1007/978-94-007-0434-3
Nikoh, N., Hosokawa, T., Moriyama, M., Oshima, K., Hattori, M., and Fukatsu, T. (2014). Evolutionary origin of insect-Wolbachia nutritional mutualism. Proc. Natl. Acad. Sci. 111, 10257–10262. doi: 10.1073/pnas.1409284111
Noel, G. R., and Atibalentja, N. (2006). “Candidatus Paenicardinium endonii” an endosymbiont of the plant-parasitic nematode Heterodera glycines (Nemata: tylenchida), affiliated to the phylum bacteroidetes. Int. J. Syst. Evol. Microbiol. 56, 1697–1702. doi: 10.1099/ijs.0.64234-0
Nováková, E., and Moran, N. A. (2012). Diversification of genes for carotenoid biosynthesis in aphids following an ancient transfer from a fungus. Mol. Biol. Evol. 29, 313–323. doi: 10.1093/molbev/msr206
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). MetaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Palomares-Rius, J. E., Archidona-Yuste, A., Cantalapiedra-Navarrete, C., Prieto, P., and Castillo, P. (2016). Molecular diversity of bacterial endosymbionts associated with dagger nematodes of the genus Xiphinema (Nematoda: Longidoridae) reveals a high degree of phylogenetic congruence with their host. Mol. Ecol. 25, 6225–6247. doi: 10.1111/mec.13904
Palomares-Rius, J. E., Gutiérrez-Gutiérrez, C., Mota, M., Bert, W., Claeys, M., Yushin, V. V., et al. (2021). Candidatus Xiphinematincola pachtaicus” gen. Nov., sp. nov., an endosymbiotic bacterium associated with nematode species of the genus Xiphinema (Nematoda, Longidoridae). Int. J. Syst. Evol. Microbiol 71:004888. doi: 10.1099/ijsem.0.004888
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pratelli, R., and Pilot, G. (2014). Regulation of amino acid metabolic enzymes and transporters in plants. J. Exp. Bot. 65, 5535–5556. doi: 10.1093/jxb/eru320
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Rao, A. U., Carta, L. K., Lesuisse, E., and Hamza, I. (2005). Lack of heme synthesis in a free-living eukaryote. Proc. Natl. Acad. Sci. U.S.A. 102, 4270–4275. doi: 10.1073/pnas.0500877102
Renvoisé, A., Merhej, V., Georgiades, K., and Raoult, D. (2011). Intracellular rickettsiales: insights into manipulators of eukaryotic cells. Trends Mol. Med. 17, 573–583. doi: 10.1016/j.molmed.2011.05.009
Rodionov, D. A., Vitreschak, A. G., Mironov, A. A., and Gelfand, M. S. (2003). Regulation of lysine biosynthesis and transport genes in bacteria: yet another RNA riboswitch? Nucleic Acids Res. 31, 6748–6757. doi: 10.1093/nar/gkg900
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rota-Stabelli, O., Daley, A. C., and Pisani, D. (2013). Molecular timetrees reveal a cambrian colonization of land and a new scenario for ecdysozoan evolution. Curr. Biol. 23, 392–398. doi: 10.1016/j.cub.2013.01.026
Scholz, M., Albanese, D., Tuohy, K., Donati, C., Segata, N., and Rota-Stabelli, O. (2020). Large scale genome reconstructions illuminate Wolbachia evolution. Nat. Commun 11:5235. doi: 10.1038/s41467-020-19016-0
Seaver, S. M. D., Liu, F., Zhang, Q., Jeffryes, J., Faria, J. P., Edirisinghe, J. N., et al. (2021). The ModelSEED biochemistry database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 49, D575–D588. doi: 10.1093/nar/gkaa746
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 7, 539. doi: 10.1038/msb.2011.75
Slatko, B. E., Taylor, M. J., and Foster, J. M. (2010). The Wolbachia endosymbiont as an anti-filarial nematode target. Symbiosis 51, 55–65. doi: 10.1007/s13199-010-0067-1
Sloan, S., Jenvey, C. J., Piedrafita, D., Preston, S., and Stear, M. J. (2021). Comparative evaluation of different molecular methods for DNA extraction from individual Teladorsagia circumcincta nematodes. BMC Biotechnol. 21:35. doi: 10.1186/s12896-021-00695-6
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Taylor, M. J., and Hoerauf, A. (1999). Wolbachia bacteria of filarial nematodes. Parasitol. Today 15, 437–442. doi: 10.1016/S0169-4758(99)01533-1
Taylor, M., Mediannikov, O., Raoult, D., and Greub, G. (2012). Endosymbiotic bacteria associated with nematodes, ticks and amoebae. FEMS Immunol. Med. Microbiol. 64, 21–31. doi: 10.1111/j.1574-695X.2011.00916.x
Topalović, O., and Vestergård, M. (2021). Can microorganisms assist the survival and parasitism of plant-parasitic nematodes? Trends Parasitol. 37, 947–958. doi: 10.1016/j.pt.2021.05.007
Uribe-Alvarez, C., Chiquete-Félix, N., Morales-García, L., Bohórquez-Hernández, A., Delgado-Buenrostro, N. L., Vaca, L., et al. (2019). Wolbachia pipientis grows in Saccharomyces cerevisiae evoking early death of the host and deregulation of mitochondrial metabolism. Microbiologyopen 8, 1–16. doi: 10.1002/mbo3.675
Vandekerckhove, T. T. M., Willems, A., Gillis, M., and Coomans, A. (2000). Occurrence of novel verrucomicrobial species, endosymbiotic and associated with parthenogenesis in Xiphinema americanum-group species (Nematoda. Longidoridae). Int. J. Syst. Evol. Microbiol. 50, 2197–2205. doi: 10.1099/00207713-50-6-2197
Vieira, P., and Gleason, C. (2019). Plant-parasitic nematode effectors –insights into their diversity and new tools for their identification. Curr. Opin. Plant Biol. 50, 37–43. doi: 10.1016/j.pbi.2019.02.007
Vollmer, J., Schiefer, A., Schneider, T., Jülicher, K., Johnston, K. L., Taylor, M. J., et al. (2013). Requirement of lipid II biosynthesis for cell division in cell wall-less Wolbachia, endobacteria of arthropods and filarial nematodes. Int. J. Med. Microbiol. 303, 140–149. doi: 10.1016/j.ijmm.2013.01.002
Waeyenberge, L., de Sutter, N., Viaene, N., and Haegeman, A. (2019). New insights into nematode DNA-metabarcoding as revealed by the characterization of artificial and spiked nematode communities. Diversity 11, 1–22. doi: 10.3390/d11040052
Wasala, S. K., Brown, A. M. V., Kang, J., Howe, D. K., Peetz, A. B., Zasada, I. A., et al. (2019). Variable abundance and distribution of wolbachia and cardinium endosymbionts in plant-parasitic nematode field populations. Front. Microbiol. 10:964. doi: 10.3389/fmicb.2019.00964
Werren, J. H. (1997). Biology of Wolbachia. Annu. Rev. Entomol. 42, 587–609. doi: 10.1146/annurev.ento.42.1.587
Werren, J. H., Baldo, L., and Clark, M. E. (2008). Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751. doi: 10.1038/nrmicro1969
Wu, B., Novelli, J., Foster, J., Vaisvila, R., Conway, L., Ingram, J., et al. (2009). The heme biosynthetic pathway of the obligate Wolbachia endosymbiont of Brugia malayi as a potential anti-filarial drug target. PLoS Negl. Trop. Dis. 3:e475. doi: 10.1371/journal.pntd.0000475
Wu, B., Novelli, J., Jiang, D., Dailey, H. A., Landmann, F., Ford, L., et al. (2013). Interdomain lateral gene transfer of an essential ferrochelatase gene in human parasitic nematodes. Proc. Natl. Acad. Sci. U.S.A. 110, 7748–7753. doi: 10.1073/pnas.1304049110
Wu, M., Sun, L. V., Vamathevan, J., Riegler, M., Deboy, R., Brownlie, J. C., et al. (2004). Phylogenomics of the reproductive parasite Wolbachia pipientis wmel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2:E69. doi: 10.1371/journal.pbio.0020069
Xu, J., Han, M., Ren, X., and Zhang, W. (2016). Modification of aspartokinase III and dihydrodipicolinate synthetase increases the production of L-lysine in Escherichia coli. Biochem. Eng. J. 114, 79–86. doi: 10.1016/j.bej.2016.06.025
Yang, H., and Ludewig, U. (2014). Lysine catabolism, amino acid transport, and systemic acquired resistance: what is the link? Plant Signal. Behav. 9:e28933. doi: 10.4161/psb.28933
Yang, H., Postel, S., Kemmerling, B., and Ludewig, U. (2014). Altered growth and improved resistance of Arabidopsis against Pseudomonas syringae by overexpression of the basic amino acid transporter Atcat1. Plant, Cell Environ. 37, 1404–1414. doi: 10.1111/pce.12244
Zečić, A., Dhondt, I., and Braeckman, B. P. (2019). The nutritional requirements of Caenorhabditis elegans. Genes Nutr. 14, 1–13. doi: 10.1186/s12263-019-0637-7
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2014). PEAR: a fast and accurate Illumina Paired-end read merger. Bioinformatics 30, 614–620. doi: 10.1093/bioinformatics/btt593
Zhang, Z., Li, J., Zhao, X. Q., Wang, J., Wong, G. K. S., and Yu, J. (2006). KaKs_calculator: calculating ka and ks through model selection and model averaging. Genomics Proteomics Bioinforma 4, 259–263. doi: 10.1016/S1672-0229(07)60007-2
Zheng, F., Zhu, D., Chen, Q. L., Bi, Q. F., Yang, X. R., O’Connor, P., et al. (2020). The driving factors of nematode gut microbiota under long-term fertilization. FEMS Microbiol. Ecol. 96, 1–11. doi: 10.1093/femsec/fiaa037
Keywords: plant-parasitic nematode, Wolbachia, enrichment, phylogenomics, endosymbiont, metagenomics
Citation: Weyandt N, Aghdam SA and Brown AMV (2022) Discovery of Early-Branching Wolbachia Reveals Functional Enrichment on Horizontally Transferred Genes. Front. Microbiol. 13:867392. doi: 10.3389/fmicb.2022.867392
Received: 01 February 2022; Accepted: 24 March 2022;
Published: 25 April 2022.
Edited by:
Alex C. C. Wilson, University of Miami, United StatesReviewed by:
Rosario Gil, University of Valencia, SpainBeatriz Sabater-Munoz, Polytechnic University of Valencia, Spain
Copyright © 2022 Weyandt, Aghdam and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda M. V. Brown, amanda.mv.brown@ttu.edu