
Multi-Omics Immune Interaction Networks in Lung Cancer Tumorigenesis, Proliferation, and Survival
 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Further Validation of the Seven-Gene Signature in Prognosis for NSCLC
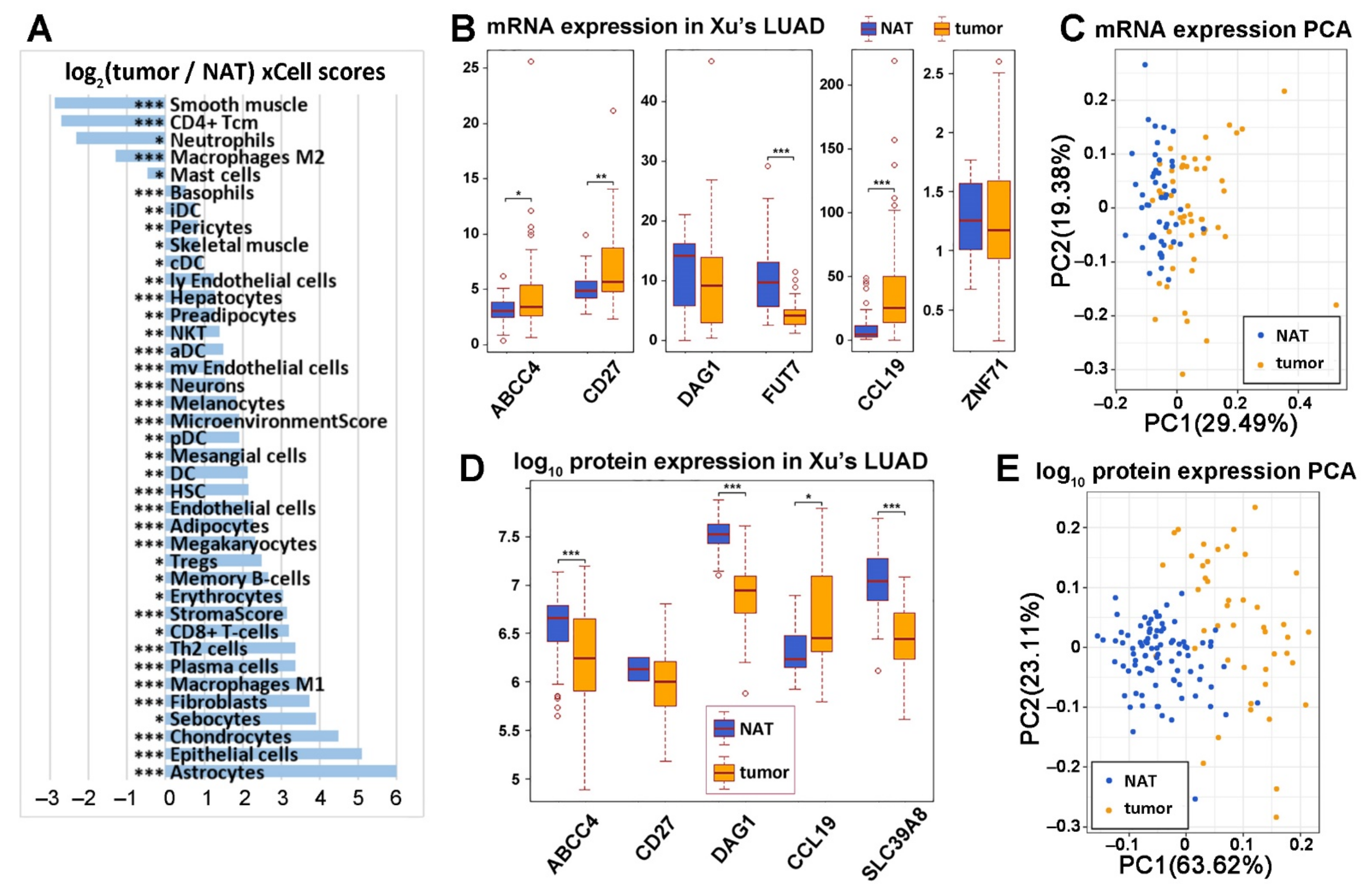
2.2. Diagnostic Implication of the Seven-Gene Signature in NSCLC
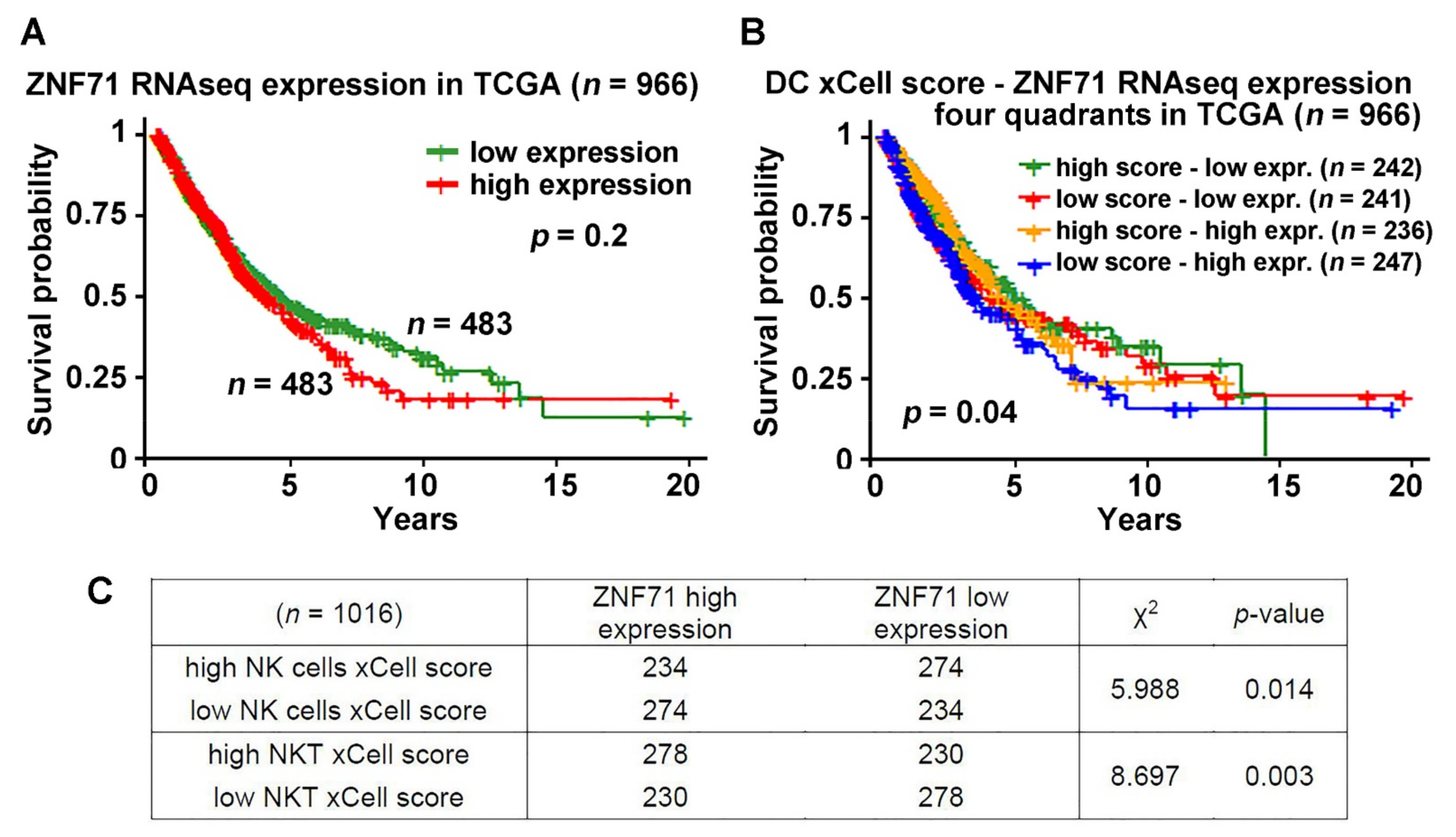
2.3. ZNF71 Expression and Selected Immune Cells in NSCLC Prognosis
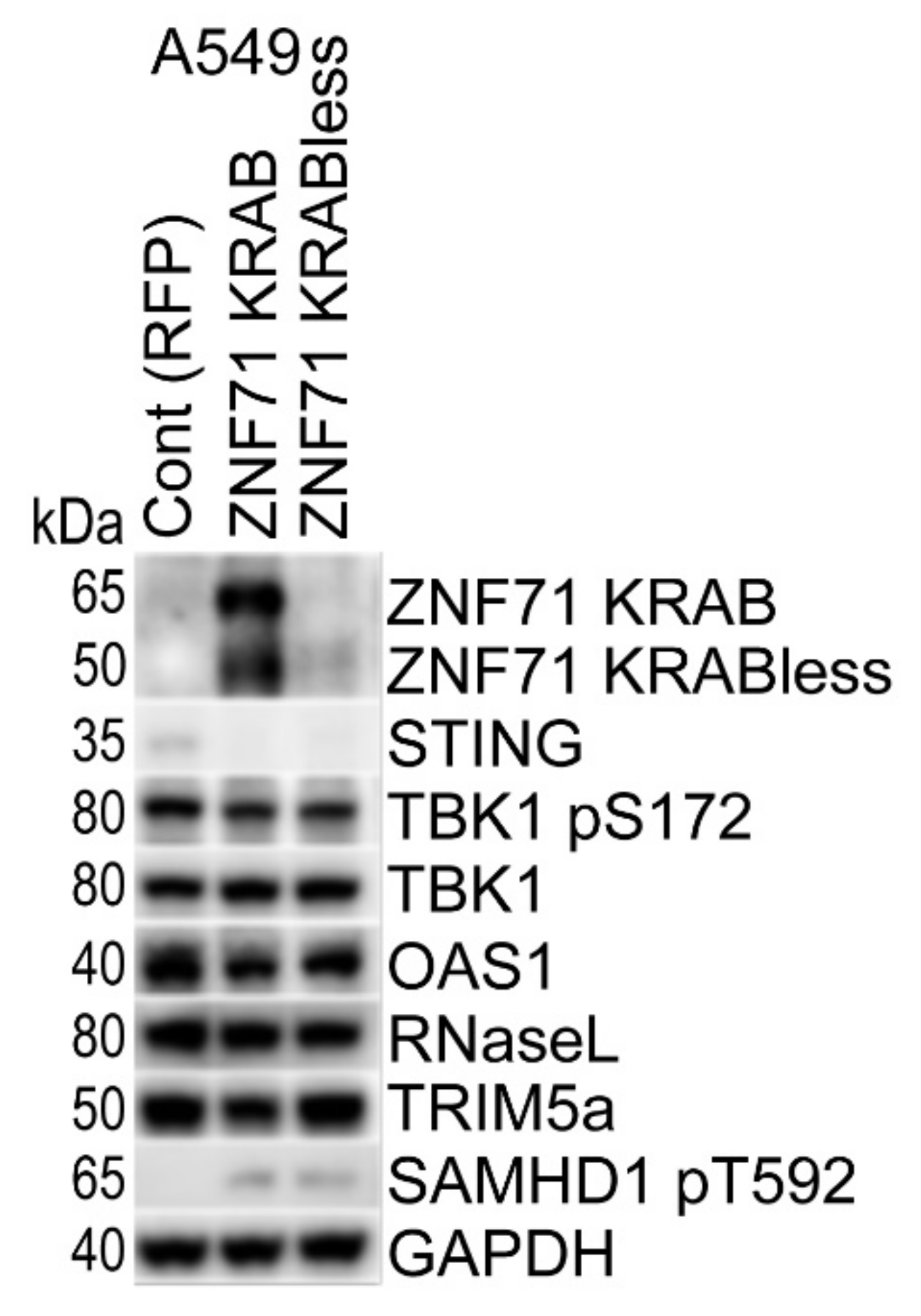
2.4. Overexpression of ZNF71 KRAB, KRAB-Less Isoforms Suppresses Innate Immune Response
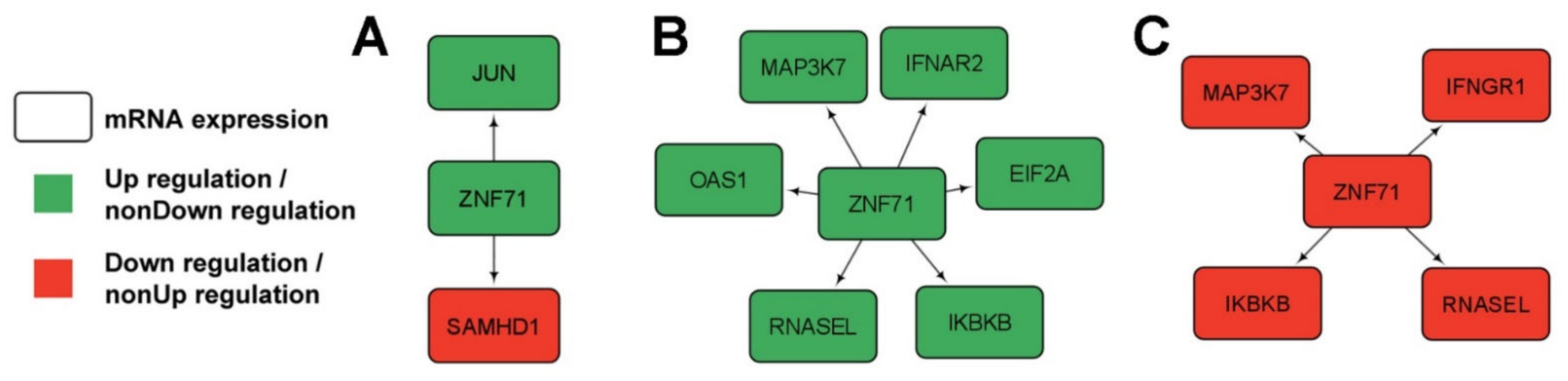
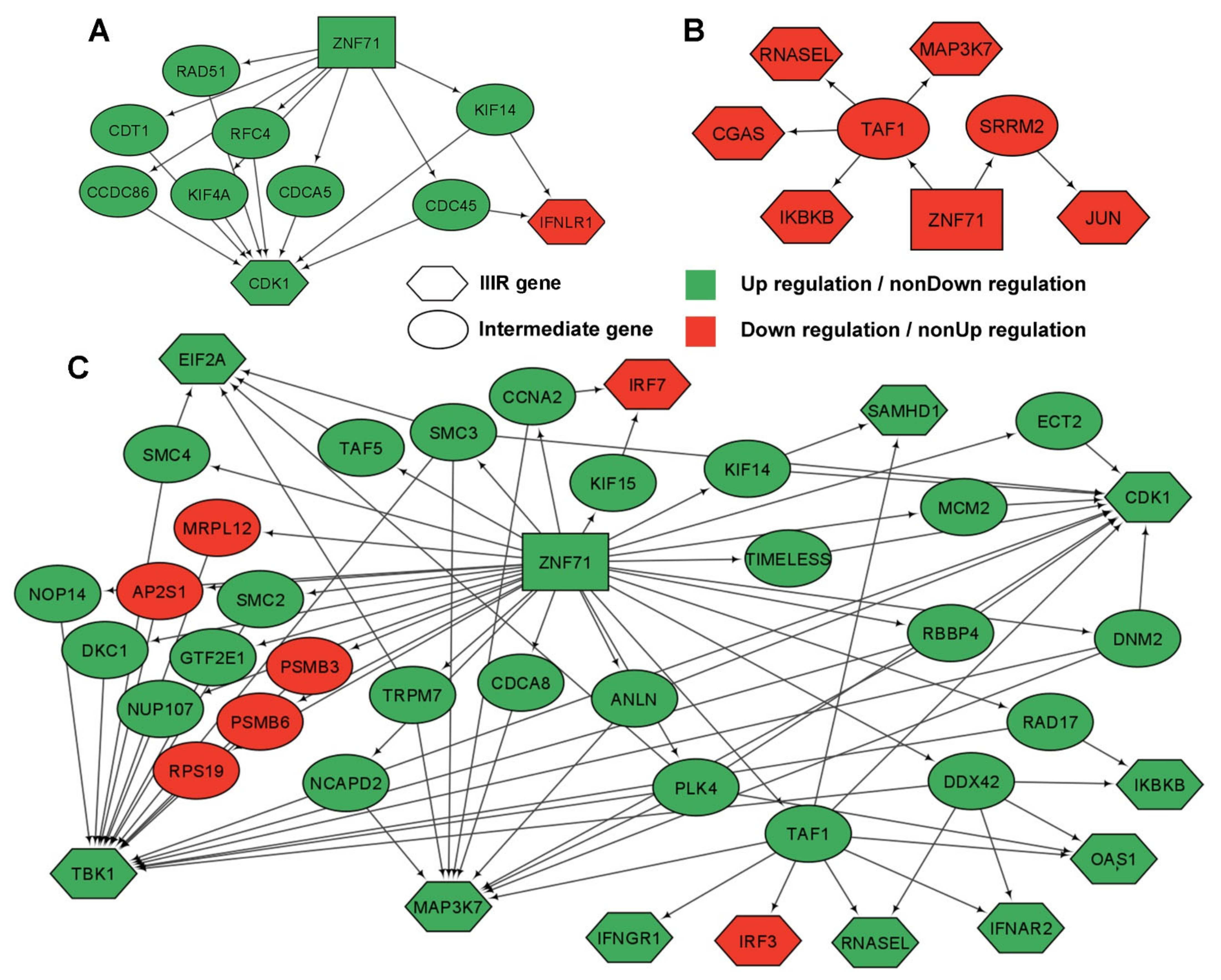
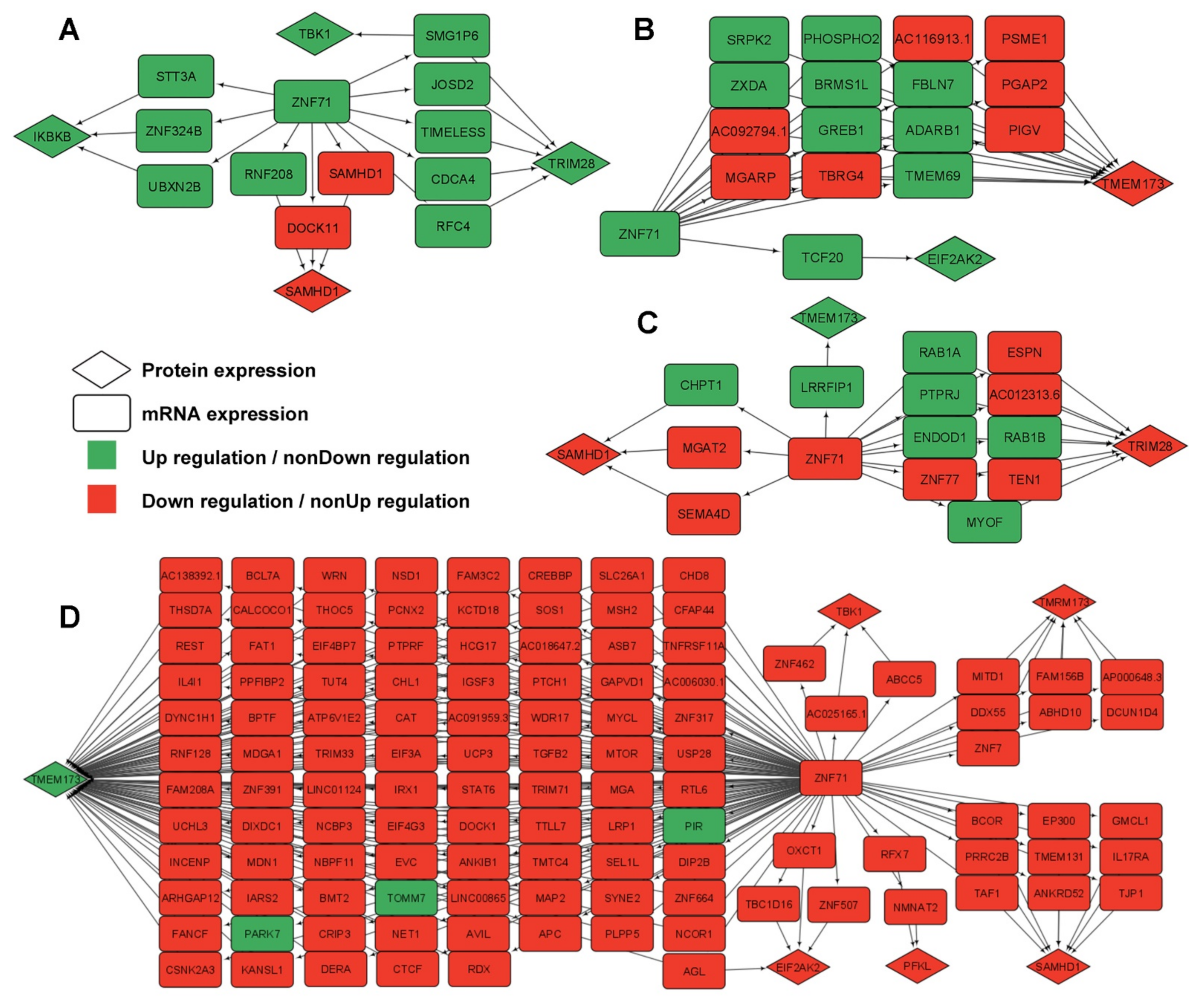
2.5. Gene Association Networks of ZNF71 and Intracellular Innate Response Genes
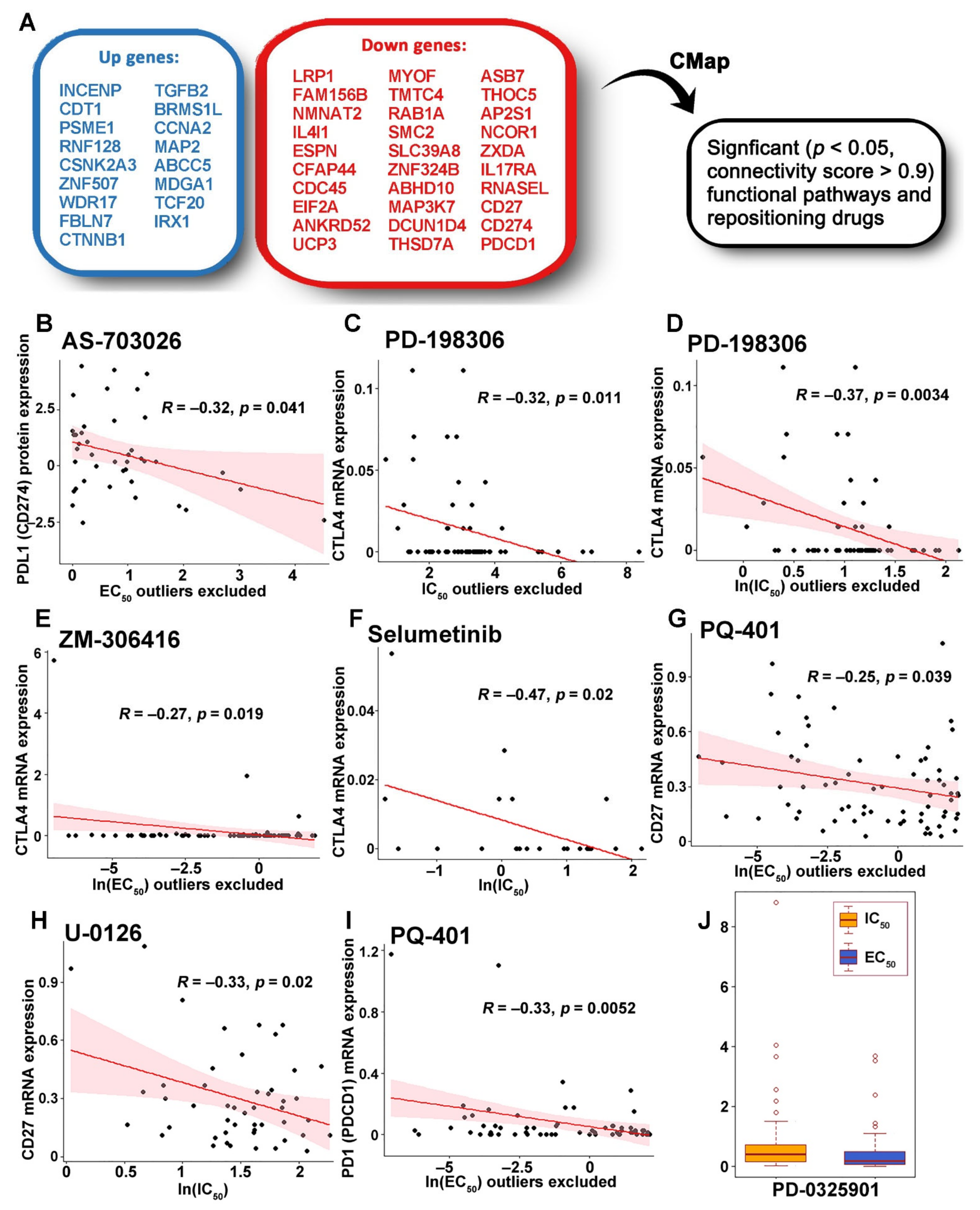
2.6. Identification of Genes Associated with Drug Response
2.7. Functional Pathways Associated with the ZNF71 Co-Expression Networks and Discovery of Therapeutic Targets
2.8. Potential Oncogenes and Tumor Suppressor Genes
3. Discussion
4. Materials and Methods
4.1. Non-Small Cell Lung Cancer (NSCLC) Patient Cohorts
4.2. xCell
4.3. Weka
4.4. Cell Lines
4.5. Vector Construction and Lentiviral Transduction
4.6. Western Blot
4.7. Boolean Implication Network
4.8. CRISPR-Cas9 Knockout Assays
4.9. RNAi Knockdown Assays
4.10. Pathway Enrichment Analysis Using ToppGene
4.11. Cancer Cell Line Encyclopedia (CCLE)
4.12. Drug Sensitivity in CCLE
4.13. Drug Repurposing Using Connectivity Map (CMap)
4.14. Statistical Methods
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lung Cancer—Non-Small Cell: Statistics. Available online: https://www.cancer.net/cancer-types/lung-cancer-non-small-cell/statistics (accessed on 23 May 2022).
- Ho, C.; Tong, K.M.; Ramsden, K.; Ionescu, D.N.; Laskin, J. Histologic classification of non-small-cell lung cancer over time: Reducing the rates of not-otherwise-specified. Curr. Oncol. 2015, 22, e164–e170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCCN Guidelines, Version: 3.2022. Available online: https://www.nccn.org/guidelines/category_1 (accessed on 28 June 2022).
- Zheng, Y.; Jaklitsch, M.T.; Bueno, R. Neoadjuvant Therapy in Non-Small Cell Lung Cancer. Surg. Oncol. Clin. N. Am. 2016, 25, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef]
- Chaft, J.E.; Rimner, A.; Weder, W.; Azzoli, C.G.; Kris, M.G.; Cascone, T. Evolution of systemic therapy for stages I-III non-metastatic non-small-cell lung cancer. Nat. Rev. Clin. Oncol. 2021, 18, 547–557. [Google Scholar] [CrossRef]
- FDA Approves Atezolizumab as Adjuvant Treatment for Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-adjuvant-treatment-non-small-cell-lung-cancer (accessed on 18 October 2021).
- Hoffman, P.C.; Mauer, A.M.; Vokes, E.E. Lung cancer. Lancet 2000, 355, 479–485. [Google Scholar] [CrossRef]
- Naruke, T.; Goya, T.; Tsuchiya, R.; Suemasu, K. Prognosis and survival in resected lung carcinoma based on the new international staging system. J. Thorac. Cardiovasc. Surg. 1988, 96, 440–447. [Google Scholar] [CrossRef]
- Vogt, U.; Striehn, E.; Bosse, U.; Klinke, F.; Falkiewicz, B. Lack of squamous cell lung carcinoma in vitro chemosensitivity to various drug regimens in the adenosine triphosphate cell viability chemosensitivity assay. Acta Biochim. Pol. 1999, 46, 299–302. [Google Scholar] [CrossRef]
- Byron, E.; Pinder-Schenck, M. Systemic and targeted therapies for early-stage lung cancer. Cancer Control 2014, 21, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Rena, O.; Oliaro, A.; Cavallo, A.; Filosso, P.L.; Donati, G.; Di Marzio, P.; Maggi, G.; Ruffini, E. Stage I non-small cell lung carcinoma: Really an early stage? Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2002, 21, 514–519. [Google Scholar] [CrossRef]
- Guo, N.L.; Dowlati, A.; Raese, R.A.; Dong, C.; Chen, G.; Beer, D.G.; Shaffer, J.; Singh, S.; Bokhary, U.; Liu, L.; et al. A Predictive 7-Gene Assay and Prognostic Protein Biomarkers for Non-small Cell Lung Cancer. EBioMedicine 2018, 32, 102–110. [Google Scholar] [CrossRef]
- Ye, Q.; Falatovich, B.; Singh, S.; Ivanov, A.V.; Eubank, T.D.; Guo, N.L. A Multi-Omics Network of a Seven-Gene Prognostic Signature for Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2021, 23, 219. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Infante, J.R.; Ansell, S.M.; Nemunaitis, J.J.; Weiss, G.R.; Villalobos, V.M.; Sikic, B.I.; Taylor, M.H.; Northfelt, D.W.; Carson, W.E., 3rd; et al. Safety and Activity of Varlilumab, a Novel and First-in-Class Agonist Anti-CD27 Antibody, in Patients With Advanced Solid Tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2028–2036. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Mohamed, R.; Dukhlallah, D.; Gencheva, M.; Hu, G.; Pearce, M.C.; Kolluri, S.K.; Marsh, C.B.; Eubank, T.D.; Ivanov, A.V.; et al. Molecular Analysis of ZNF71 KRAB in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 3752. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.; Mahgoub, M.; Macfarlan, T.S. The Arms Race Between KRAB-Zinc Finger Proteins and Endogenous Retroelements and Its Impact on Mammals. Annu. Rev. Genet. 2019, 53, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Sadeq, S.; Al-Hashimi, S.; Cusack, C.M.; Werner, A. Endogenous Double-Stranded RNA. Non-Coding RNA 2021, 7, 15. [Google Scholar] [CrossRef]
- Kassiotis, G. Endogenous retroviruses and the development of cancer. J. Immunol. 2014, 192, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef]
- Di Giorgio, E.; Xodo, L.E. Endogenous Retroviruses (ERVs): Does RLR (RIG-I-Like Receptors)-MAVS Pathway Directly Control Senescence and Aging as a Consequence of ERV De-Repression? Front. Immunol 2022, 13, 917998. [Google Scholar] [CrossRef] [PubMed]
- Hur, S. Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu. Rev. Immunol. 2019, 37, 349–375. [Google Scholar] [CrossRef]
- Yang, E.; Li, M.M.H. All about the RNA: Interferon-Stimulated Genes That Interfere With Viral RNA Processes. Front. Immunol. 2020, 11, 605024. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Kim, S.; Kim, M.; Kang, M.G.; Kwak, C.; Kang, M.; Kim, B.; Rhee, H.W.; Kim, V.N. PKR Senses Nuclear and Mitochondrial Signals by Interacting with Endogenous Double-Stranded RNAs. Mol. Cell 2018, 71, 1051–1063.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamontin, C.; Bossis, G.; Nisole, S.; Arhel, N.J.; Maarifi, G. Regulation of Viral Restriction by Post-Translational Modifications. Viruses 2021, 13, 2197. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Kishore, N.; Huynh, Q.K.; Mathialagan, S.; Hall, T.; Rouw, S.; Creely, D.; Lange, G.; Caroll, J.; Reitz, B.; Donnelly, A.; et al. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: Comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J. Biol. Chem. 2002, 277, 13840–13847. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.Y.; Zhang, C.; Wang, X.; Zhai, L.; Ma, Y.; Mao, Y.; Qian, K.; Sun, C.; Liu, Z.; Jiang, S.; et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell 2020, 182, 245–261.e17. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Woo, S.R.; Burnett, B.; Fu, Y.X.; Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013, 34, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Rizvi, N.A.; Goldman, J.W.; Gettinger, S.N.; Borghaei, H.; Brahmer, J.R.; Ready, N.E.; Gerber, D.E.; Chow, L.Q.; Juergens, R.A.; et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open-label, phase 1, multicohort study. Lancet. Oncol. 2017, 18, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Quint, L.E.; Kretschmer, M.; Chang, A.; Nan, B. CT-guided thoracic core biopsies: Value of a negative result. Cancer Imaging Off. Publ. Int. Cancer Imaging Soc. 2006, 6, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Casaluce, F.; Sgambato, A.; Maione, P.; Sacco, P.C.; Santabarbara, G.; Gridelli, C. Selumetinib for the treatment of non-small cell lung cancer. Expert Opin. Investig. Drugs 2017, 26, 973–984. [Google Scholar] [CrossRef]
- Wang, W.W.; Wang, W.Q.; Wang, S.S.; Pan, L. The efficacy and safety of selumetinib as secondary therapy for late-stage and metastatic non-small cell lung cancer: Results from a systematic review and meta-analysis. Ann. Transl. Med. 2022, 10, 593. [Google Scholar] [CrossRef]
- Lebbé, C.; Italiano, A.; Houédé, N.; Awada, A.; Aftimos, P.; Lesimple, T.; Dinulescu, M.; Schellens, J.H.M.; Leijen, S.; Rottey, S.; et al. Selective Oral MEK1/2 Inhibitor Pimasertib in Metastatic Melanoma: Antitumor Activity in a Phase I, Dose-Escalation Trial. Target. Oncol. 2021, 16, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Delord, J.P.; Italiano, A.; Awada, A.; Aftimos, P.; Houédé, N.; Lebbé, C.; Pages, C.; Lesimple, T.; Dinulescu, M.; Schellens, J.H.M.; et al. Selective Oral MEK1/2 Inhibitor Pimasertib: A Phase I Trial in Patients with Advanced Solid Tumors. Target. Oncol. 2021, 16, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Ripple, M.O.; Kim, N.; Springett, R. Acute mitochondrial inhibition by mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK) 1/2 inhibitors regulates proliferation. J. Biol. Chem. 2013, 288, 2933–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besson, B.; Kim, S.; Kim, T.; Ko, Y.; Lee, S.; Larrous, F.; Song, J.; Shum, D.; Grailhe, R.; Bourhy, H. Kinome-Wide RNA Interference Screening Identifies Mitogen-Activated Protein Kinases and Phosphatidylinositol Metabolism as Key Factors for Rabies Virus Infection. mSphere 2019, 4, e00047-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, J.M.; Carrasco, G.A. Cannabinoid receptor agonists upregulate and enhance serotonin 2A (5-HT(2A)) receptor activity via ERK1/2 signaling. Synapse 2013, 67, 145–159. [Google Scholar] [CrossRef] [Green Version]
- Ciruela, A.; Dixon, A.K.; Bramwell, S.; Gonzalez, M.I.; Pinnock, R.D.; Lee, K. Identification of MEK1 as a novel target for the treatment of neuropathic pain. Br. J. Pharmacol. 2003, 138, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Aksamitiene, E.; Kholodenko, B.N.; Kolch, W.; Hoek, J.B.; Kiyatkin, A. PI3K/Akt-sensitive MEK-independent compensatory circuit of ERK activation in ER-positive PI3K-mutant T47D breast cancer cells. Cell. Signal. 2010, 22, 1369–1378. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, J.P.; Fernandes, J.C.; Brunet, J.; Moldovan, F.; Schrier, D.; Flory, C.; Martel-Pelletier, J. In vivo selective inhibition of mitogen-activated protein kinase kinase 1/2 in rabbit experimental osteoarthritis is associated with a reduction in the development of structural changes. Arthritis Rheum. 2003, 48, 1582–1593. [Google Scholar] [CrossRef]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef] [Green Version]
- Fukazawa, H.; Noguchi, K.; Murakami, Y.; Uehara, Y. Mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK) inhibitors restore anoikis sensitivity in human breast cancer cell lines with a constitutively activated extracellular-regulated kinase (ERK) pathway. Mol. Cancer 2002, 1, 303–309. [Google Scholar]
- Antczak, C.; Mahida, J.P.; Bhinder, B.; Calder, P.A.; Djaballah, H. A high-content biosensor-based screen identifies cell-permeable activators and inhibitors of EGFR function: Implications in drug discovery. J. Biomol. Screen 2012, 17, 885–899. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.B.; Xu, Y.Y.; Cheng, W.W.; Yuan, B.; Zhao, J.R.; Wang, Y.L.; Zhang, H.J. Decreased PGF may contribute to trophoblast dysfunction in fetal growth restriction. Reproduction 2017, 154, 319–329. [Google Scholar] [CrossRef]
- Zhou, X.; Zhao, X.; Li, X.; Ping, G.; Pei, S.; Chen, M.; Wang, Z.; Zhou, W.; Jin, B. PQ401, an IGF-1R inhibitor, induces apoptosis and inhibits growth, proliferation and migration of glioma cells. J. Chemother. 2016, 28, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; Zhang, R.; Sun, R.; Guo, M.; Zhang, M.; Wei, G.; Zhang, L.; Yu, S.; Huang, H. IGF-1R inhibitor PQ401 inhibits osteosarcoma cell proliferation, migration and colony formation. Int. J. Clin. Exp. Pathol. 2019, 12, 1589–1598. [Google Scholar] [PubMed]
- Haura, E.B.; Ricart, A.D.; Larson, T.G.; Stella, P.J.; Bazhenova, L.; Miller, V.A.; Cohen, R.B.; Eisenberg, P.D.; Selaru, P.; Wilner, K.D.; et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 2450–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, P.; Dera, A.; Abdalsamad, M.R.; Chandramoorthy, H.C. Rational combinations of indirubin and arylidene derivatives exhibit synergism in human non-small cell lung carcinoma cells. J. Food Biochem. 2019, 43, e12861. [Google Scholar] [CrossRef]
- Laurie, S.A.; Goss, G.D.; Shepherd, F.A.; Reaume, M.N.; Nicholas, G.; Philip, L.; Wang, L.; Schwock, J.; Hirsh, V.; Oza, A.; et al. A phase II trial of saracatinib, an inhibitor of src kinases, in previously-treated advanced non-small-cell lung cancer: The princess margaret hospital phase II consortium. Clin. Lung Cancer 2014, 15, 52–57. [Google Scholar] [CrossRef]
- Franks, S.E.; Jones, R.A.; Briah, R.; Murray, P.; Moorehead, R.A. BMS-754807 is cytotoxic to non-small cell lung cancer cells and enhances the effects of platinum chemotherapeutics in the human lung cancer cell line A549. BMC Res. Notes 2016, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Xu, L.F.; Zhang, J.; Kong, S.Y.; Wu, M.; Lao, Y.Z.; Zhou, H.; Zhang, L.; Xu, H. SRC and MEK Co-inhibition Synergistically Enhances the Anti-tumor Effect in Both Non-small-cell Lung Cancer (NSCLC) and Erlotinib-Resistant NSCLC. Front. Oncol. 2019, 9, 586. [Google Scholar] [CrossRef] [Green Version]
- Ciuleanu, T.E.; Ahmed, S.; Kim, J.H.; Mezger, J.; Park, K.; Thomas, M.; Chen, J.; Poondru, S.; VanTornout, J.M.; Whitcomb, D.; et al. Randomised Phase 2 study of maintenance linsitinib (OSI-906) in combination with erlotinib compared with placebo plus erlotinib after platinum-based chemotherapy in patients with advanced non-small cell lung cancer. Br. J. Cancer 2017, 117, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Leighl, N.B.; Rizvi, N.A.; de Lima, L.G., Jr.; Arpornwirat, W.; Rudin, C.M.; Chiappori, A.A.; Ahn, M.J.; Chow, L.Q.; Bazhenova, L.; Dechaphunkul, A.; et al. Phase 2 Study of Erlotinib in Combination With Linsitinib (OSI-906) or Placebo in Chemotherapy-Naive Patients With Non-Small-Cell Lung Cancer and Activating Epidermal Growth Factor Receptor Mutations. Clin. Lung Cancer 2017, 18, 34–42.e2. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yang, J.; Yang, C.; Huang, X.; Han, M.; Kang, F.; Li, J. Morphine promotes the malignant biological behavior of non-small cell lung cancer cells through the MOR/Src/mTOR pathway. Cancer Cell Int. 2021, 21, 622. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Ramos-Alvarez, I.; Moreno, P.; Mantey, S.A.; Ridnour, L.; Wink, D.; Jensen, R.T. Endothelin causes transactivation of the EGFR and HER2 in non-small cell lung cancer cells. Peptides 2017, 90, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suehiro, Y.; Okada, T.; Shikamoto, N.; Zhan, Y.; Sakai, K.; Okayama, N.; Nishioka, M.; Furuya, T.; Oga, A.; Kawauchi, S.; et al. Germline copy number variations associated with breast cancer susceptibility in a Japanese population. Tumour Biol. 2013, 34, 947–952. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.C.; Lee, D.F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 2004, 117, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Splittgerber, R.; Yull, F.E.; Kantrow, S.; Ayers, G.D.; Karin, M.; Richmond, A. Conditional ablation of Ikkb inhibits melanoma tumor development in mice. J. Clin. Investig. 2010, 120, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Zhang, J.; Tang, B.; Jing, L.; Qiu, B.; Zha, Z. The novel circ_0028171/miR-218-5p/IKBKB axis promotes osteosarcoma cancer progression. Cancer Cell Int. 2020, 20, 484. [Google Scholar] [CrossRef]
- Bowers, J.L.; Randell, J.C.; Chen, S.; Bell, S.P. ATP hydrolysis by ORC catalyzes reiterative Mcm2-7 assembly at a defined origin of replication. Mol. Cell 2004, 16, 967–978. [Google Scholar] [CrossRef]
- Forsburg, S.L. Eukaryotic MCM proteins: Beyond replication initiation. Microbiol. Mol. Biol. Rev. MMBR 2004, 68, 109–131. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Lei, C.; Pan, B.; Fang, S.; Chen, Y.; Cao, W.; Liu, L. Potential Prospective Biomarkers for Non-small Cell Lung Cancer: Mini-Chromosome Maintenance Proteins. Front. Genet. 2021, 12, 587017. [Google Scholar] [CrossRef]
- Yuan, J.; Lan, H.; Huang, D.; Guo, X.; Liu, C.; Liu, S.; Zhang, P.; Cheng, Y.; Xiao, S. Multi-Omics Analysis of MCM2 as a Promising Biomarker in Pan-Cancer. Front. Cell Dev. Biol. 2022, 10, 852135. [Google Scholar] [CrossRef]
- Xiang, J.; Fang, L.; Luo, Y.; Yang, Z.; Liao, Y.; Cui, J.; Huang, M.; Yang, Z.; Huang, Y.; Fan, X.; et al. Levels of human replication factor C4, a clamp loader, correlate with tumor progression and predict the prognosis for colorectal cancer. J. Transl. Med. 2014, 12, 320. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Yang, Q.; Wei, B.; Yuan, B.; Liu, Y.; Li, Y.; Gu, D.; Yin, G.; Wang, B.; Xu, D.; et al. Investigation of crucial genes and microRNAs in conventional osteosarcoma using gene expression profiling analysis. Mol. Med. Rep. 2017, 16, 7617–7624. [Google Scholar] [CrossRef] [PubMed]
- Aryal, R.P.; Kwak, P.B.; Tamayo, A.G.; Gebert, M.; Chiu, P.L.; Walz, T.; Weitz, C.J. Macromolecular Assemblies of the Mammalian Circadian Clock. Mol. Cell 2017, 67, 770–782.e6. [Google Scholar] [CrossRef] [Green Version]
- Onyeisi, J.O.S.; Lopez, C.C.; Götte, M. Role of Syndecan-4 in breast cancer pathophysiology. Am. J. Physiol. Cell Physiol. 2022, 323, C1345. [Google Scholar] [CrossRef]
- Miller, M.R.; Ma, D.; Schappet, J.; Breheny, P.; Mott, S.L.; Bannick, N.; Askeland, E.; Brown, J.; Henry, M.D. Downregulation of dystroglycan glycosyltransferases LARGE2 and ISPD associate with increased mortality in clear cell renal cell carcinoma. Mol. Cancer 2015, 14, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, B.W.; Lathia, J.D.; Bruce, Z.C.; D’Souza, R.C.J.; Baumgartner, U.; Ensbey, K.S.; Lim, Y.C.; Stringer, B.W.; Akgul, S.; Offenhauser, C.; et al. The dystroglycan receptor maintains glioma stem cells in the vascular niche. Acta Neuropathol. 2019, 138, 1033–1052. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Hou, Y.; Hu, J.; Zhou, L.; Chen, K.; Yang, X.; Song, Z. SLC39A8/Zinc Suppresses the Progression of Clear Cell Renal Cell Carcinoma. Front. Oncol. 2021, 11, 651921. [Google Scholar] [CrossRef]
- An, X.; Zhu, Y.; Zheng, T.; Wang, G.; Zhang, M.; Li, J.; Ji, H.; Li, S.; Yang, S.; Xu, D.; et al. An Analysis of the Expression and Association with Immune Cell Infiltration of the cGAS/STING Pathway in Pan-Cancer. Mol. Ther. Nucleic Acids 2019, 14, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ma, M.; Li, L.; Huang, Y.; Zhao, G.; Zhou, Y.; Yang, Y.; Yang, Y.; Wang, B.; Ye, L. MTA1-TJP1 interaction and its involvement in non-small cell lung cancer metastasis. Transl. Oncol. 2022, 25, 101500. [Google Scholar] [CrossRef]
- Moon, S.U.; Kim, J.H.; Woo, H.G. Tumor suppressor RBM24 inhibits nuclear translocation of CTNNB1 and TP63 expression in liver cancer cells. Oncol. Lett. 2021, 22, 674. [Google Scholar] [CrossRef]
- Wu, H.; Lu, X.X.; Wang, J.R.; Yang, T.Y.; Li, X.M.; He, X.S.; Li, Y.; Ye, W.L.; Wu, Y.; Gan, W.J.; et al. TRAF6 inhibits colorectal cancer metastasis through regulating selective autophagic CTNNB1/β-catenin degradation and is targeted for GSK3B/GSK3β-mediated phosphorylation and degradation. Autophagy 2019, 15, 1506–1522. [Google Scholar] [CrossRef]
- Katoh, M. Multi-layered prevention and treatment of chronic inflammation, organ fibrosis and cancer associated with canonical WNT/β-catenin signaling activation (Review). Int. J. Mol. Med. 2018, 42, 713–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas, M.A.; Meydan, C.; Chin, C.R.; Challman, M.F.; Kim, D.; Bhinder, B.; Kloetgen, A.; Viny, A.D.; Teater, M.R.; McNally, D.R.; et al. Smc3 dosage regulates B cell transit through germinal centers and restricts their malignant transformation. Nat. Immunol. 2021, 22, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Okabe, C.; Fujii, K.; Kato, Y.; Ogihara, T. Regulation of breast cancer resistance protein and P-glycoprotein by ezrin, radixin and moesin in lung, intestinal and renal cancer cell lines. J. Pharm. Pharmacol. 2020, 72, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software. ACM SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Addison, J.B.; Voronkova, M.A.; Fugett, J.H.; Lin, C.C.; Linville, N.C.; Trinh, B.; Livengood, R.H.; Smolkin, M.B.; Schaller, M.D.; Ruppert, J.M.; et al. Functional hierarchy and cooperation of EMT master transcription factors in breast cancer metastasis. Mol. Cancer Res. 2021, 19, 784–798. [Google Scholar] [CrossRef]
- Guo, L.; Cukic, B.; Singh, H. Predicting Fault Prone Modules by the Dempster-Shafer Belief Networks. In Proceedings of the 18th IEEE International Conference on Automated Software Engineering (ASE’03), Montreal, QC, Canada, 6–10 October 2003; pp. 249–252. [Google Scholar]
- Guo, N.L.; Wan, Y.W.; Bose, S.; Denvir, J.; Kashon, M.L.; Andrew, M.E. A novel network model identified a 13-gene lung cancer prognostic signature. Int. J. Comput. Biol. Drug Des. 2011, 4, 19–39. [Google Scholar] [CrossRef]
- Guo, N.L.; Wan, Y.W. Pathway-based identification of a smoking associated 6-gene signature predictive of lung cancer risk and survival. Artif. Intell. Med. 2012, 55, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Singh, S.; Qian, P.R.; Guo, N.L. Immune-Omics Networks of CD27, PD1, and PDL1 in Non-Small Cell Lung Cancer. Cancers 2021, 13, 4296. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.F.; Werner, R.; Vollbrecht, C.; Hager, T.; Flom, E.; Christoph, D.C.; Schmeller, J.; Schmid, K.W.; Wohlschlaeger, J.; Mairinger, F.D. ACTB, CDKN1B, GAPDH, GRB2, RHOA and SDCBP Were Identified as Reference Genes in Neuroendocrine Lung Cancer via the nCounter Technology. PLoS ONE 2016, 11, e0165181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saviozzi, S.; Cordero, F.; Lo Iacono, M.; Novello, S.; Scagliotti, G.V.; Calogero, R.A. Selection of suitable reference genes for accurate normalization of gene expression profile studies in non-small cell lung cancer. BMC Cancer 2006, 6, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.C.; Ding, Y.; Dong, L.; Zhu, L.J.; Jensen, R.V.; Hsiao, L.L. Differential expression patterns of housekeeping genes increase diagnostic and prognostic value in lung cancer. PeerJ 2018, 6, e4719. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Dempster, J.M.R.J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019, 720243. [Google Scholar] [CrossRef]
- DepMap, Broad. DepMap 21Q4 Public. In Figshare [Internet]. 2021. Available online: https://figshare.com/articles/dataset/DepMap_21Q4_Public/16924132/1 (accessed on 24 November 2022).
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef] [Green Version]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jané-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402.e16. [Google Scholar] [CrossRef]
- R Team. RStudio: Integrated Development Environment for R, Version 1.4.1106; RStudio, PBC.: Boston, MA, USA, 2020. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Systemic/Targeted Therapy | Pan-Sensitive Genes | Pan-Resistant Genes |
|---|---|---|---|
| afatinib | EGFR Exon 19 Deletion or L858R/EGFR S768I, L861Q, and/or G719X | CDT1, INCENP | IL4I1, LRP1, FAM156B, DCUN1D4, ESPN, PTPRJ, NMNAT2 |
| alectinib | ALK Rearrangement Positive | CSNK2A3, DYNC1H1, PSME1 | UCP3, CDC45, THSD7A, ANKRD52, EIF2A, FAM156B, KRT8, CFAP44 |
| brigatinib | ALK Rearrangement Positive | MYOF, ABHD10, TMTC4 | |
| cabozantinib | RET Rearrangement Positive | CSNK2A3 | RNASEL, TMTC4, SMC2, ASB7, CFAP44, IL17RA, ZXDA, THSD7A |
| carboplatin | Systemic | ASB7, RAB1A | |
| cisplatin | Systemic | ZNF507, CSNK2A3, WDR17 | ARHGAP12, CDC45, ESPN, EIF2A, IL17RA, MYOF, TMTC4, NMNAT2, PTPRJ, KRT8 |
| crizotinib | ALK Rearrangement Positive/ROS1 Rearrangement Positive/MET Exon 14 Skipping Mutation | RFC4, CSNK2A3, MCM2, MDN1, CDT1, INCENP | LRP1, IL17RA |
| dabrafenib | BRAF V600E Mutation Positive | FBLN7, MAP2, CTNNB1, RNF128, CSNK2A3, WDR17, ZNF507 | FAM156B, SLC39A8, SMC2, KRT8, NMNAT2 |
| dacomitinib | EGFR Exon 19 Deletion or L858R/EGFR S768I, L861Q, and/or G719X | CDT1 | MAP3K7, DCUN1D4, LRP1, THOC5, FAM156B, ANKRD52, THSD7A |
| docetaxel | Systemic | RFC4, ABCC5, FBLN7, MAP2 | SLC39A8, IL17RA, KRT8, RAB1A, THSD7A, DCUN1D4, MYOF, NMNAT2 |
| erlotinib | EGFR Exon 19 Deletion or L858R/EGFR S768I, L861Q, and/or G719X | CSNK2A3, PSMB6, TGFB2 | FAM156B, ASB7, TMTC4, NMNAT2 |
| etoposide | Systemic | PSMB3, CDT1, DYNC1H1, ZNF507 | ESPN, NMNAT2, ZNF324B, IL17RA |
| gefitinib | EGFR Exon 19 Deletion or L858R/EGFR S768I, L861Q, and/or G719X | BRMS1L, CCNA2, CDT1, INCENP | IL4I1, ABHD10, FAM156B, MAP3K7, DCUN1D4, NMNAT2, PTPRJ, KRT8 |
| gemcitabine | Systemic | MDGA1, MDN1, ZNF507, WDR17 | KRT8, ANKRD52, ASB7, RAB1A, FAM156B, IL17RA, DCUN1D4, IL4I1, LRP1, NMNAT2 |
| lorlatinib | ALK Rearrangement Positive/ROS1 Rearrangement Positive | MDN1, PSMB6 | ANKRD52, ESPN, RAB1A |
| osimertinib | EGFR Exon 19 Deletion or L858R/EGFR S768I, L861Q, and/or G719X | ABCC5, MDGA1, TCF20, MAP2, PSMB6 | ASB7, DCUN1D4, THSD7A |
| paclitaxel | Systemic | INCENP, CDT1, MCM2, TCF20 | RAB1A, THOC5, ASB7, IL17RA, NMNAT2, KRT8, LRP1, SLC39A8, THSD7A, UCP3 |
| pemetrexed | Systemic | MCM2, CCNA2 | EIF2A, IL17RA, MYOF, THSD7A, ZXDA, ARHGAP12, LRP1 |
| trametinib | BRAF V600E Mutation Positive | CTNNB1 | RNASEL, IL17RA, ANKRD52, FAM156B, AP2S1, ARHGAP12, SRRM2, UCP3, ZXDA, TMTC4, DCUN1D4, NCOR1, RAB1A |
| vemurafenib | BRAF V600E Mutation Positive | RNASE L, IL17RA | |
| vinorelbine | Systemic | ABCC5, DYNC1H1, IRX1, TCF20 | RAB1A, SLC39A8, KRT8, DCUN1D4, PTPRJ, IL17RA, MYOF, NMNAT2 |
| Drug | Systemic/Targeted Therapy | Pan-Sensitive Genes | Pan-Resistant Genes |
|---|---|---|---|
| afatinib | EGFR Exon 19 Deletion or L858R/EGFR S768I, L861Q, and/or G719X | DDX42, RFX7, FAT1, CREBBP | IL4I1, KRT8, CDCA4, DIXDC1 |
| alectinib | ALK Rearrangement Positive | DDX42, ZNF507 | CCNA2, KIF14, CDK1, DAG1, ANKRD52, MAP3K7 |
| brigatinib | ALK Rearrangement Positive | FAT1, PTPRJ, PSME1 | TEN1, MAP3K7, FBLN7, CDK1 |
| cabozantinib | RET Rearrangement Positive | KANSL1, CHL1, TGFB2 | TJP1_G3V1L9, BPTF_E9PE19, NSD1, SYNE2_Q8WXH0_5, RDX, ANKRD52 |
| carboplatin | Systemic | TJP1_G3V1L9 | |
| cisplatin | Systemic | MYCL, DDX42, BRMS1L, KANSL1, ZNF507 | BPTF_E9PE19, FBLN7, DIP2B, MYOF, CDK1 |
| crizotinib | ALK Rearrangement Positive/ROS1 Rearrangement Positive/MET Exon 14 Skipping Mutation | FAM208A, TOMM7, KANSL1 | DAG1 |
| dabrafenib | BRAF V600E Mutation Positive | BRMS1L, RBBP4, ZNF507, DDX42, KANSL1 | RDX, ANKRD52, DIP2B, CDK1, DAG1, IL17RA |
| dacomitinib | EGFR Exon 19 Deletion or L858R/EGFR S768I, L861Q, and/or G719X | PSME1, DDX42, GREB1, RBBP4 | MAP2_P11137_4, KRT8, CDC45, CDCA4, IL4I1 |
| docetaxel | Systemic | TOMM7, CREBBP, TCF20, DDX42, FAM208A, THOC5, ZNF507 | DYNC1H1, MYOF |
| erlotinib | EGFR Exon 19 Deletion or L858R/EGFR S768I, L861Q, and/or G719X | RFX7, THOC5, CREBBP | KIF14, CDCA4 |
| etoposide | Systemic | CHL1, FAT1 | |
| gefitinib | EGFR Exon 19 Deletion or L858R/EGFR S768I, L861Q, and/or G719X | DDX42, CREBBP, TCF20, CHL1 | CDCA4, IL4I1 |
| gemcitabine | Systemic | DDX42, CREBBP, ZNF507, RBBP4, TOMM7 | DIP2B, ANKRD52, CDK1, MAP2_P11137_4, IL17RA, SLC39A8 |
| lorlatinib | ALK Rearrangement Positive/ROS1 Rearrangement Positive | ASB7, PSME1, PIR | CCNA2, MYOF, CDK1, CDC45 |
| osimertinib | EGFR Exon 19 Deletion or L858R/EGFR S768I, L861Q, and/or G719X | CDCA4, EIF4G3_B1AN89, IL4I1, ADARB1, CDC45 | |
| paclitaxel | Systemic | CHL1, THOC5, ZNF507, UCHL3 | SLC39A8, DIXDC1, DIP2B, MYOF, IL17RA |
| pemetrexed | Systemic | THOC5 | BPTF_E9PE19, RDX, DYNC1H1 |
| trametinib | BRAF V600E Mutation Positive | CHL1, PSME1, TMTC4, JOSD2, PTPRJ, RPS19 | RDX, DAG1, IL17RA, MAP3K7 |
| vemurafenib | BRAF V600E Mutation Positive | FAM208A | KRT8 |
| vinorelbine | Systemic | TCF20, UCHL3, RBBP4, THOC5, ZNF507 | TJP1_G3V1L9 |
| Src_Set_Id | Compound | Average of IC50 | Count of IC50 (Count of IC50 Outliers) | Average of EC50 | Count of EC50 (Count of EC50 Outliers) |
|---|---|---|---|---|---|
| CP_MEK_INHIBITOR | PD-0325901 | 0.748 | 64 (0) | 0.386 | 88 (0) |
| CP_SRC_INHIBITOR | dasatinib | 0.555 | 92 (0) | 0.504 | 155 (4) |
| MAP_KINASE_INHIBITOR | PD-98059 | 4.977 | 4 (0) | 0.607 | 56 (3) |
| LEUCINE_RICH_REPEAT_KINASE_INHIBITOR | indirubin | 3.548 | 23 (0) | 0.630 | 160 (7) |
| CP_MEK_INHIBITOR | selumetinib | 2.214 | 35 (0) | 0.647 | 165 (7) |
| CP_MEK_INHIBITOR | AS-703026 | 1.514 | 100 (2) | 0.940 | 164 (8) |
| CP_SRC_INHIBITOR | saracatinib | 1.733 | 103 (0) | 0.983 | 170 (6) |
| CP_IGF_1_INHIBITOR | BMS-754807 | 1.744 | 97 (0) | 1.022 | 168 (8) |
| CP_SRC_INHIBITOR | ZM-306416 | 2.626 | 28 (0) | 1.081 | 87 (1) |
| CP_SRC_INHIBITOR | bosutinib | 3.162 | 46 (0) | 1.587 | 87 (0) |
| CP_MEK_INHIBITOR | U-0126 | 4.533 | 77 (0) | 1.868 | 177 (3) |
| CP_MEK_INHIBITOR | PD-198306 | 3.164 | 67 (1) | 2.034 | 92 (0) |
| CP_SRC_INHIBITOR | PP-1 | 3.408 | 51 (0) | 2.079 | 87 (0) |
| CP_IGF_1_INHIBITOR | linsitinib | 4.251 | 64 (1) | 2.088 | 146 (9) |
| CP_MEK_INHIBITOR | PD-184352 | 3.988 | 47 (1) | 2.190 | 89 (0) |
| CP_MEK_INHIBITOR | MEK1-2-inhibitor | 4.197 | 44 (7) | 2.200 | 84 (1) |
| IGF-1_INHIBITOR | PQ-401 | 5.344 | 30 (0) | 2.204 | 79 (1) |
| CP_SRC_INHIBITOR | PP-2 | 3.187 | 64 (0) | 2.484 | 79 (1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Q.; Hickey, J.; Summers, K.; Falatovich, B.; Gencheva, M.; Eubank, T.D.; Ivanov, A.V.; Guo, N.L. Multi-Omics Immune Interaction Networks in Lung Cancer Tumorigenesis, Proliferation, and Survival. Int. J. Mol. Sci. 2022, 23, 14978. https://doi.org/10.3390/ijms232314978
Ye Q, Hickey J, Summers K, Falatovich B, Gencheva M, Eubank TD, Ivanov AV, Guo NL. Multi-Omics Immune Interaction Networks in Lung Cancer Tumorigenesis, Proliferation, and Survival. International Journal of Molecular Sciences. 2022; 23(23):14978. https://doi.org/10.3390/ijms232314978
Chicago/Turabian StyleYe, Qing, Justin Hickey, Kathleen Summers, Brianne Falatovich, Marieta Gencheva, Timothy D. Eubank, Alexey V. Ivanov, and Nancy Lan Guo. 2022. "Multi-Omics Immune Interaction Networks in Lung Cancer Tumorigenesis, Proliferation, and Survival" International Journal of Molecular Sciences 23, no. 23: 14978. https://doi.org/10.3390/ijms232314978