
A Multi-Omics Network of a Seven-Gene Prognostic Signature for Non-Small Cell Lung Cancer

 , and
, and
Abstract
:1. Introduction
2. Results
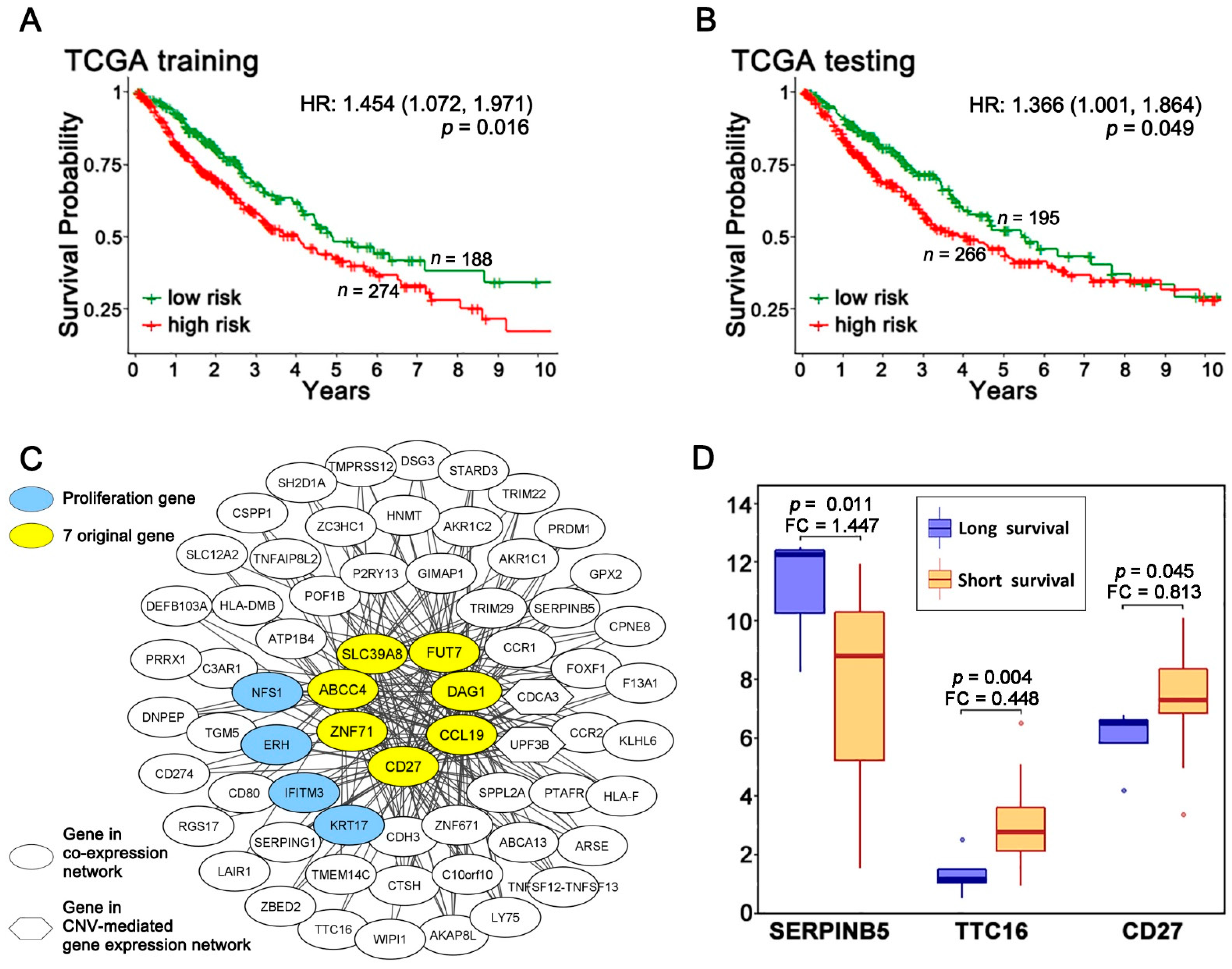
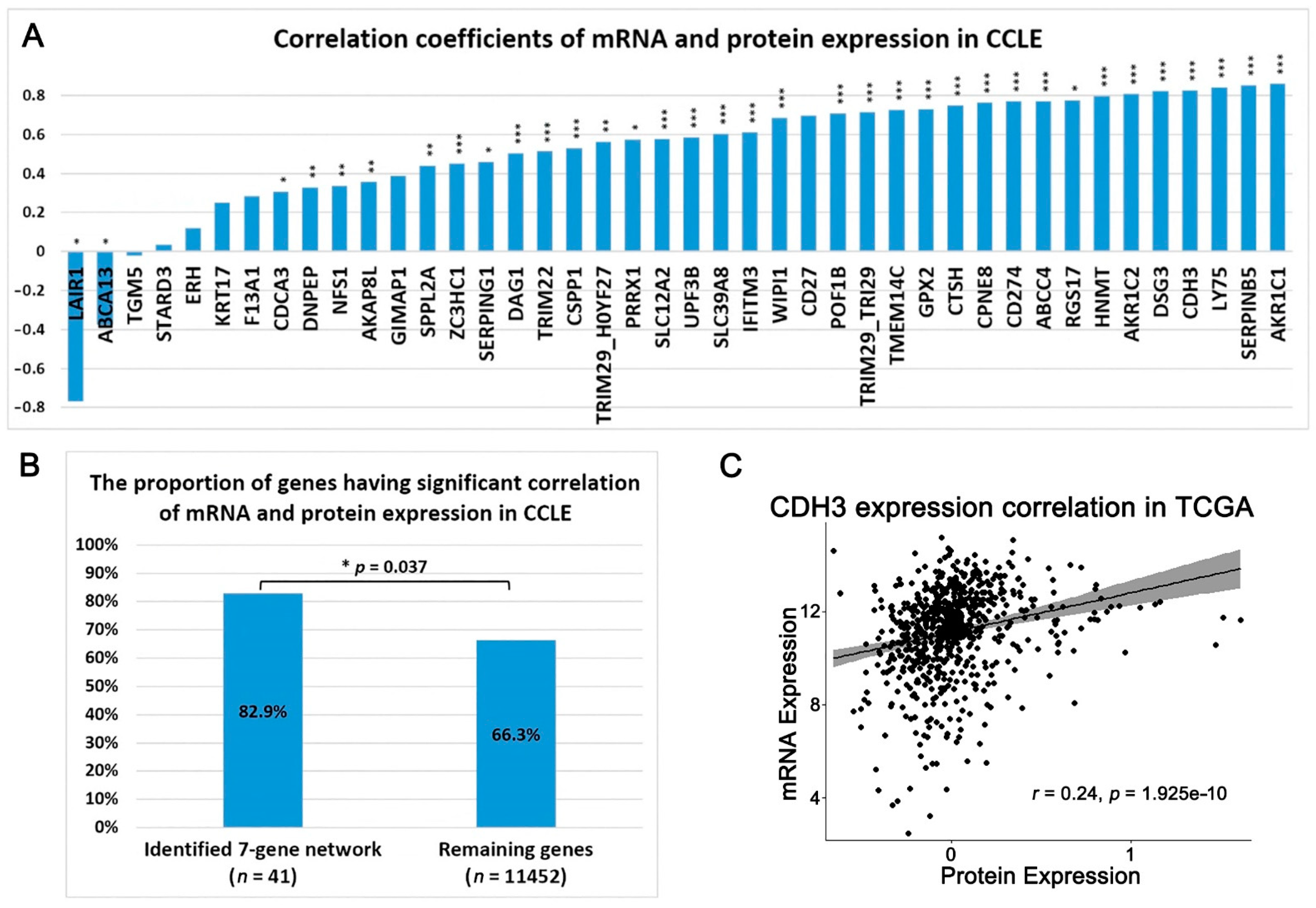
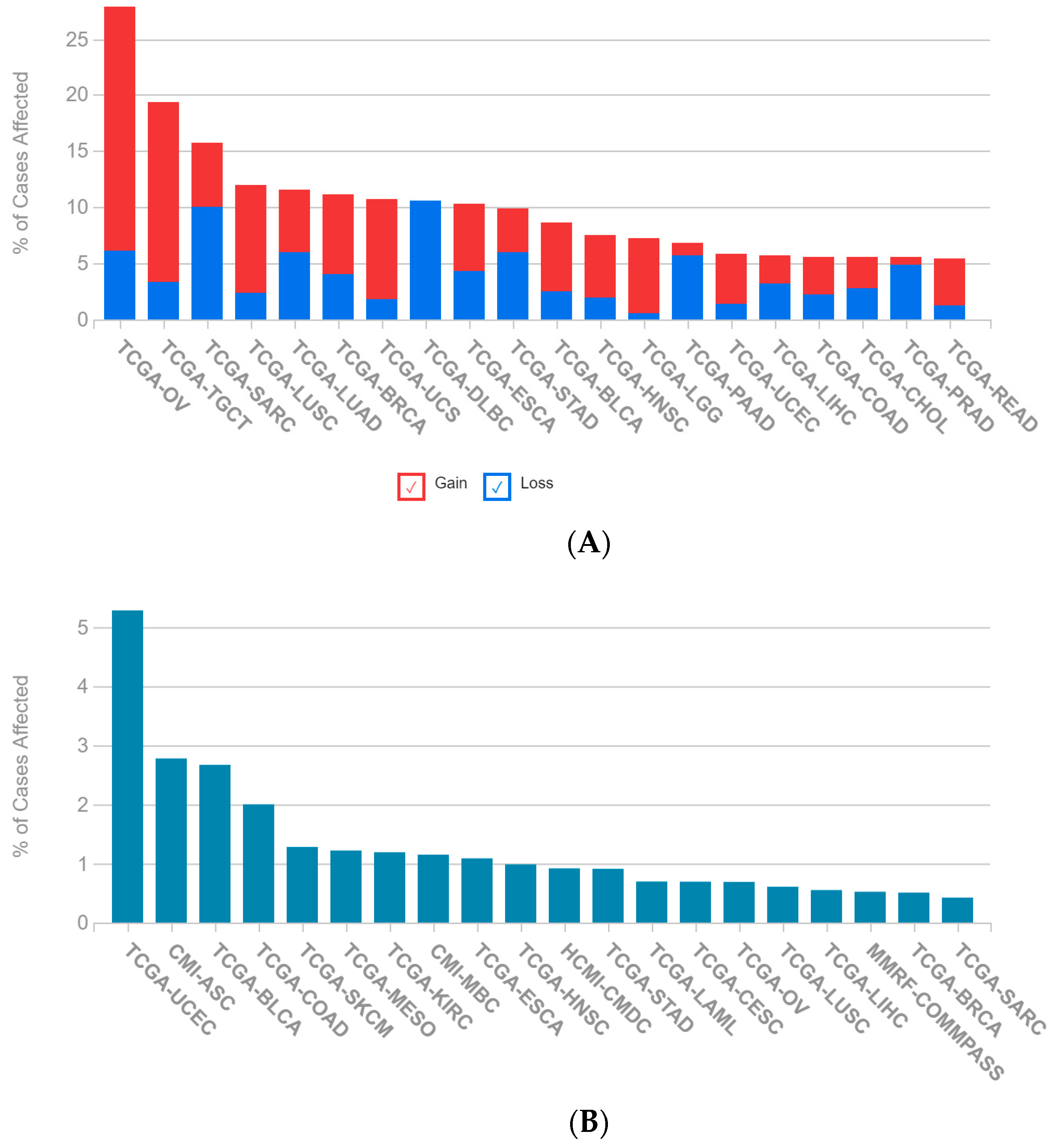
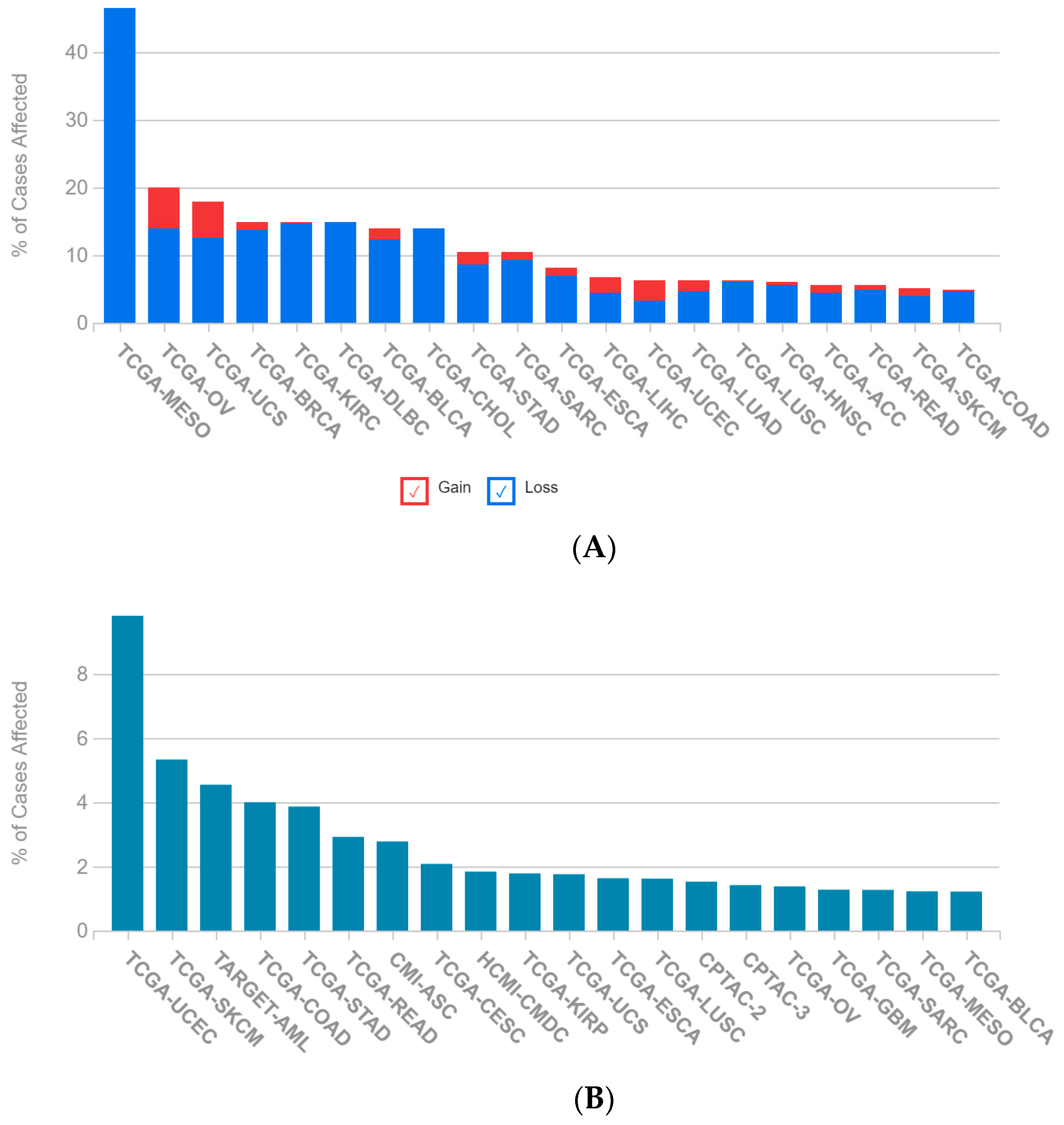
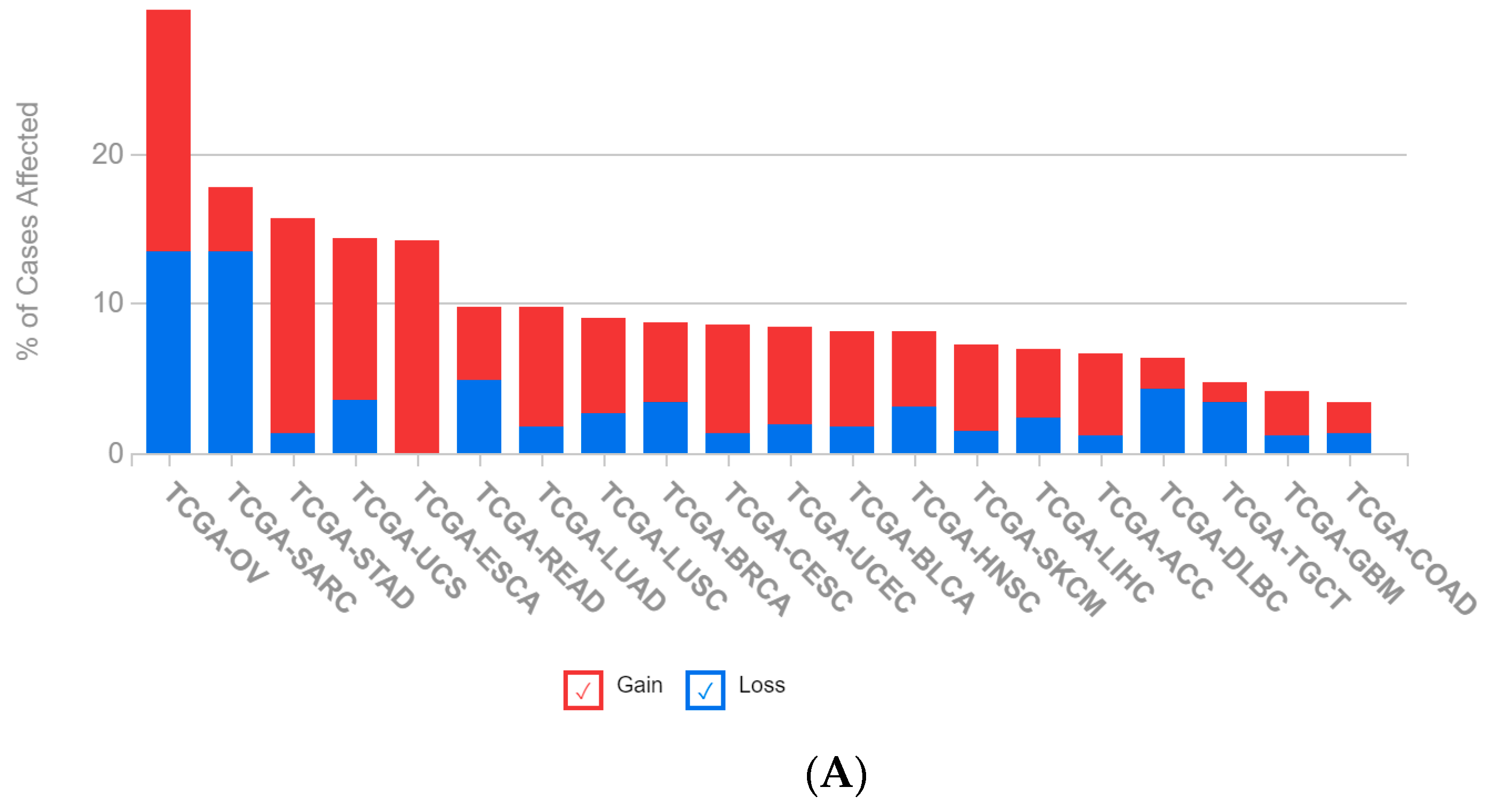
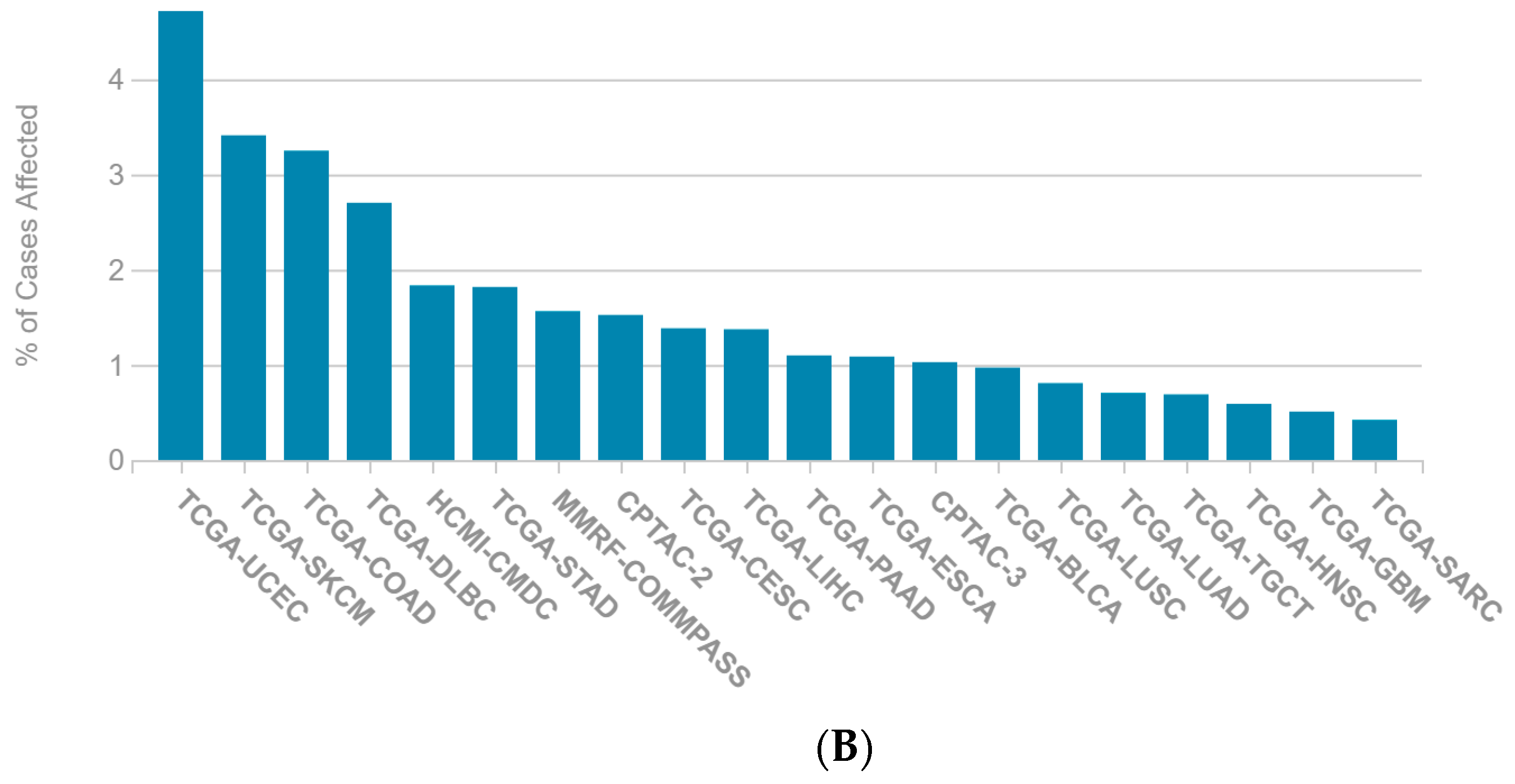
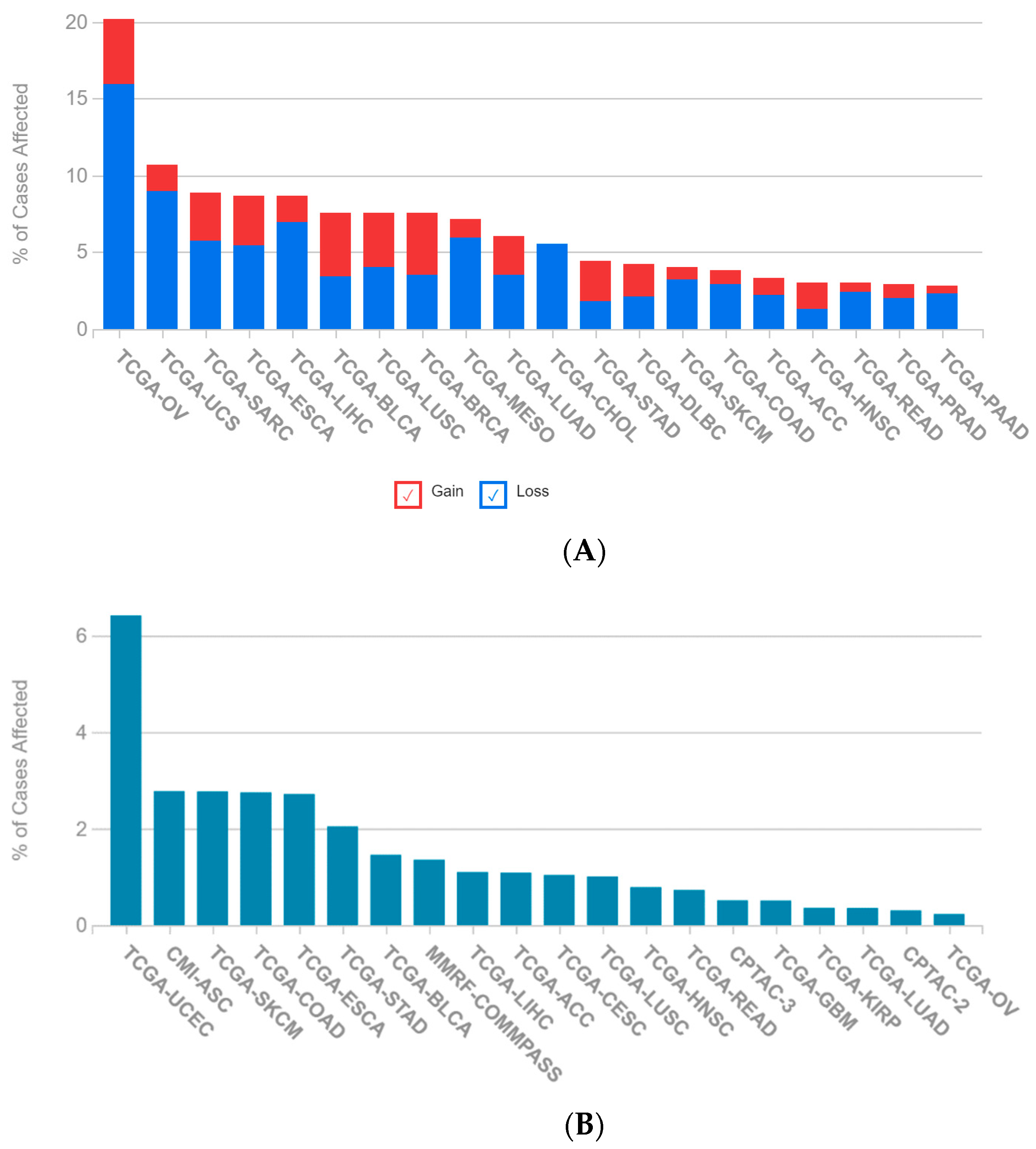
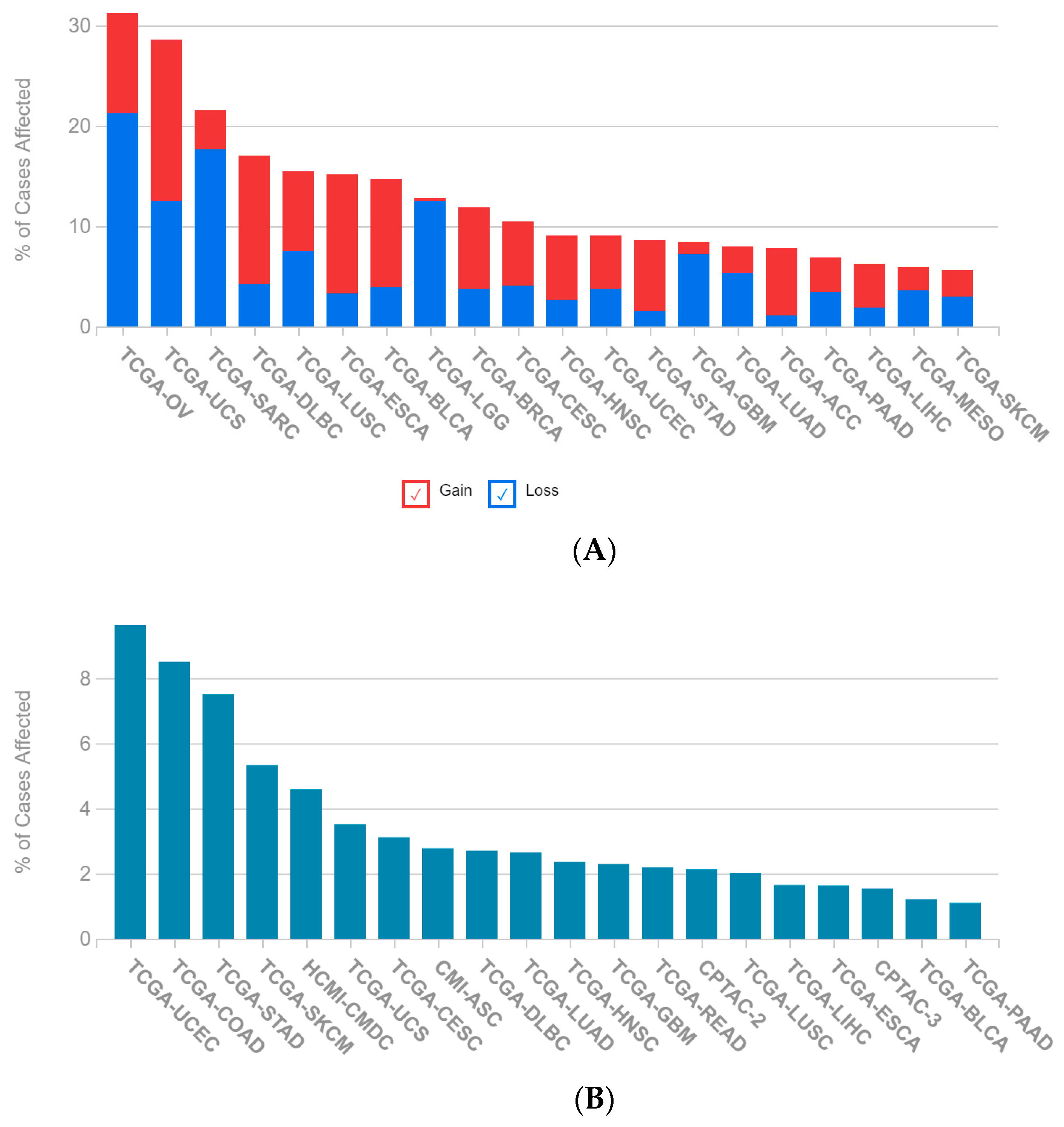
2.1. A Multi-Omics Network of the Seven-Gene NSCLC Prognostic and Predictive Signature
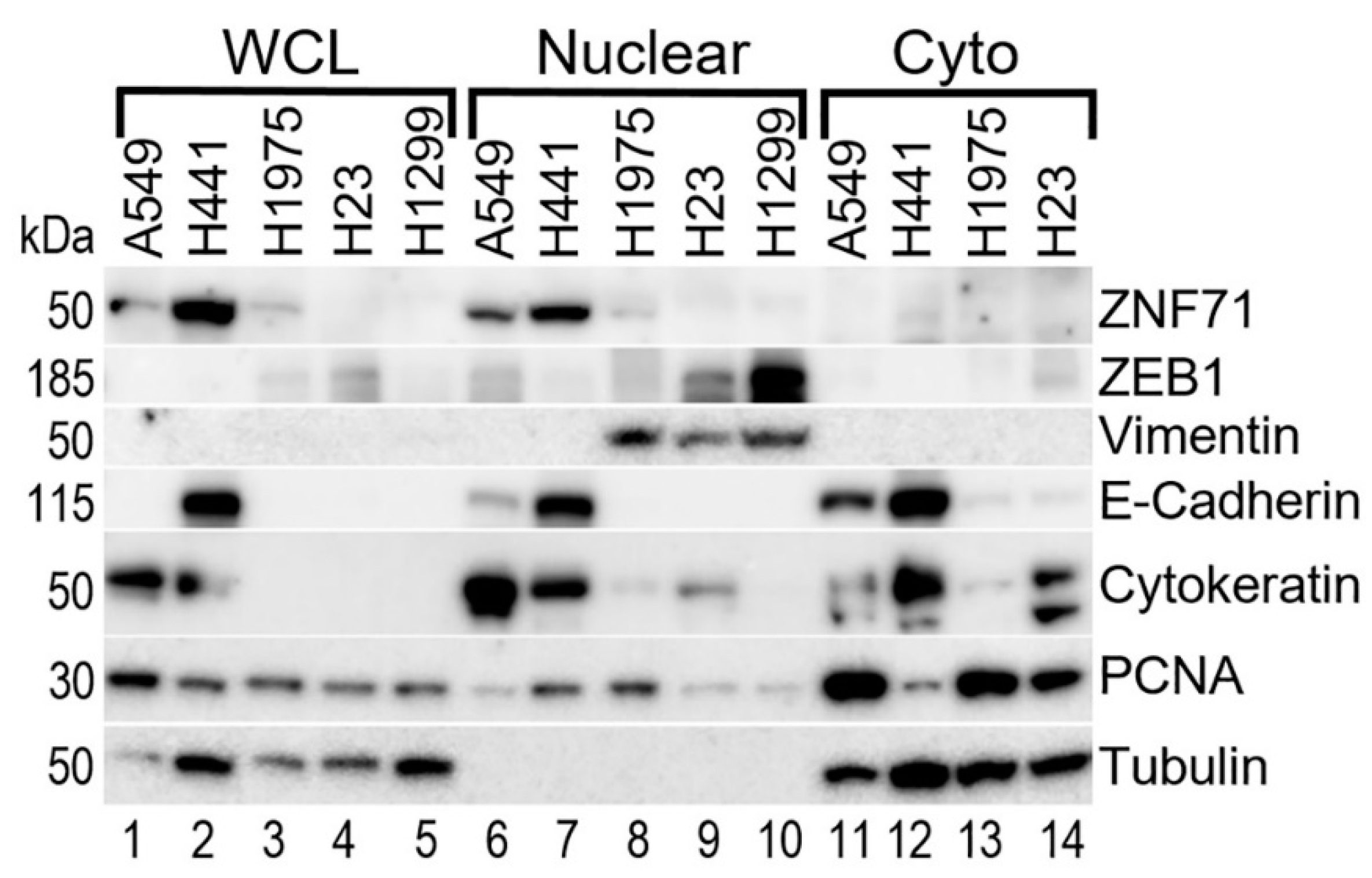
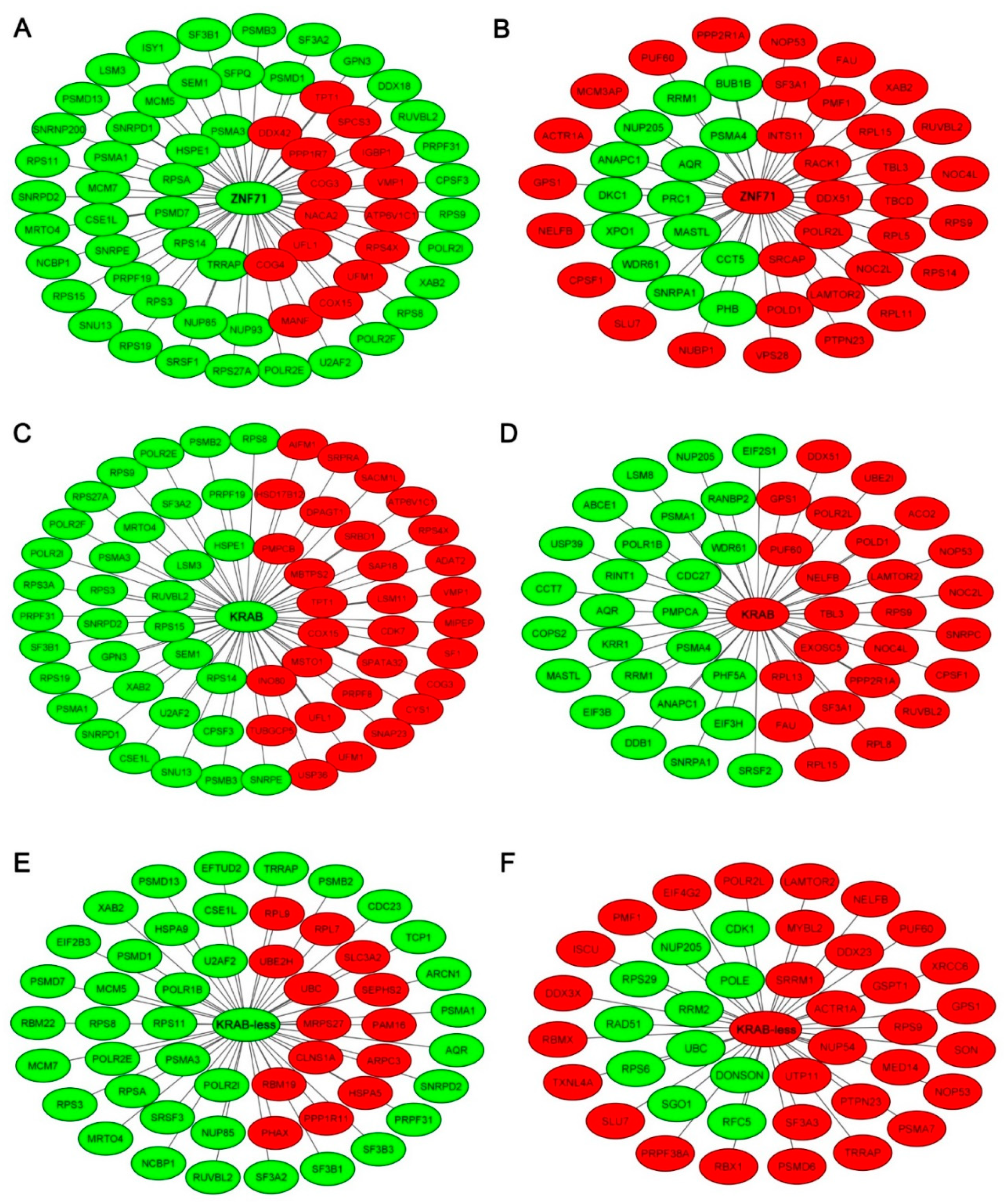
2.2. Gene Co-Expression Networks of ZNF71 and Isoforms
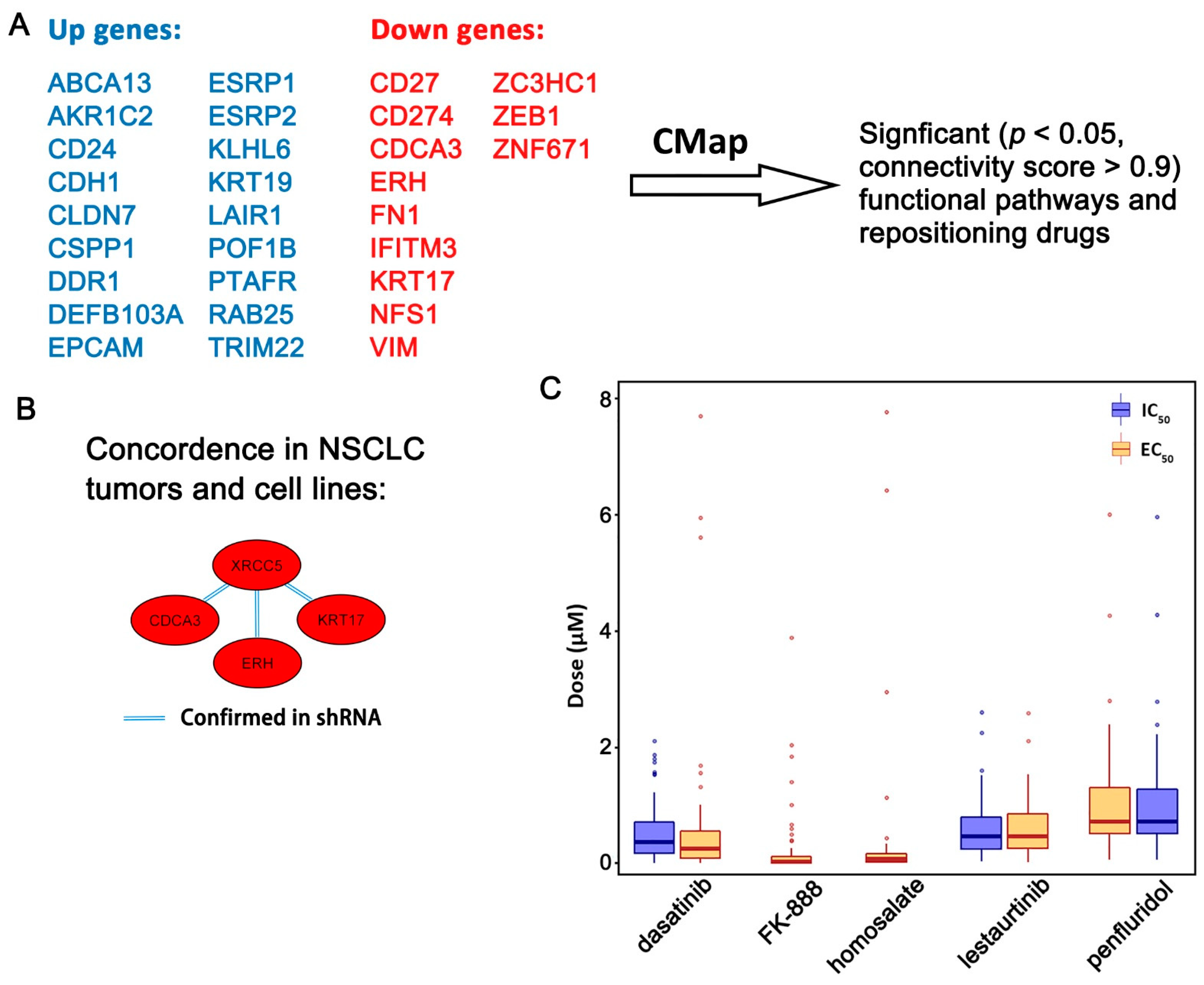
2.3. Functional Pathways Associated with the Seven-Gene Multi-Omics Network and Discovery of Repositioning Drugs
3. Discussion
4. Materials and Methods
4.1. Patient Cohorts
4.1.1. TCGA NSCLC Patient Cohorts
4.1.2. NSCLC Patient Cohort GSE31800
4.1.3. NSCLC Patient Cohort GSE28582
4.1.4. NSCLC Patient Cohort GSE81089
4.2. Data Pre-Processing
4.2.1. CNV Data Pre-Processing
4.2.2. Gene Expression Data Pre-Processing
4.3. Boolean Implication Networks
4.4. Cancer Cell Line Encyclopedia (CCLE)
4.5. CRISPR-Cas9 Knockout Assays
4.6. RNAi Knockdown Assays
4.7. Immune Infiltration Estimation
4.8. PRISM Drug Response in CCLE
4.9. Genomics of Drug Sensitivity in Cancer (GDSC1/2)
4.10. Drug Repurposing Using Connectivity Map (CMap)
4.11. Western Blots
4.12. Statistical Methods
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A









References
- Zhang, W.; Laborde, P.M.; Coombes, K.R.; Berry, D.A.; Hamilton, S.R. Cancer genomics: Promises and complexities. Clin. Cancer Res. 2001, 7, 2159–2167. [Google Scholar]
- Mahmoud, S. Non-Smokers Are at Increasing Risk for Lung Cancer. Available online: https://www.mdlinx.com/article/non-smokers-are-at-increasing-risk-for-lung-cancer/lfc-3757 (accessed on 19 September 2021).
- Spira, A.; Wood, A.J.J.; Ettinger, D.S. Multidisciplinary Management of Lung Cancer. N. Eng. J. Med. 2004, 350, 379–392. [Google Scholar] [CrossRef]
- Kim, H.S.; Mitsudomi, T.; Soo, R.A.; Cho, B.C. Personalized therapy on the horizon for squamous cell carcinoma of the lung. Lung Cancer 2013, 80, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. Molecular histology of lung cancer: From targets to treatments. Cancer Treat. Rev. 2015, 41, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jaklitsch, M.T.; Bueno, R. Neoadjuvant Therapy in Non-Small Cell Lung Cancer. Surg. Oncol. Clin. N. Am. 2016, 25, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Crino, L.; Weder, W.; van Meerbeeck, J.; Felip, E.; Group, E.G.W. Early stage and locally advanced (non-metastatic) non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21, v103–v115. [Google Scholar] [CrossRef]
- Byron, E.; Pinder-Schenck, M. Systemic and Targeted Therapies for Early-Stage Lung Cancer. Cancer Control 2014, 21, 21–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA Approves Atezolizumab as Adjuvant Treatment for Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-adjuvant-treatment-non-small-cell-lung-cancer (accessed on 8 October 2021).
- Guo, N.L.; Dowlati, A.; Raese, R.A.; Dong, C.; Chen, G.; Beer, D.G.; Shaffer, J.; Singh, S.; Bokhary, U.; Liu, L.; et al. A Predictive 7-Gene Assay and Prognostic Protein Biomarkers for Non-small Cell Lung Cancer. EBioMedicine 2018, 32, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Huntley, S.; Baggott, D.M.; Hamilton, A.T.; Tran-Gyamfi, M.; Yang, S.; Kim, J.; Gordon, L.; Branscomb, E.; Stubbs, L. A comprehensive catalog of human KRAB-associated zinc finger genes: Insights into the evolutionary history of a large family of transcriptional repressors. Genome Res. 2006, 16, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Bruno, M.; Mahgoub, M.; Macfarlan, T.S. The Arms Race Between KRAB-Zinc Finger Proteins and Endogenous Retroelements and Its Impact on Mammals. Ann. Rev. Genet. 2019, 53, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef] [Green Version]
- Lupo, A.; Cesaro, E.; Montano, G.; Zurlo, D.; Izzo, P.; Costanzo, P. KRAB-Zinc Finger Proteins: A Repressor Family Displaying Multiple Biological Functions. Curr. Genom. 2013, 14, 268–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, A.; de Herreros, A.G. Epithelial-mesenchymal transition in cancer. Mol. Oncol. 2017, 11, 715–717. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Mohamed, R.; Dukhlallah, D.; Gencheva, M.; Hu, G.; Pearce, M.C.; Kolluri, S.K.; Marsh, C.B.; Eubank, T.D.; Ivanov, A.V.; et al. Molecular Analysis of ZNF71 KRAB in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 3752. [Google Scholar] [CrossRef]
- Chen, J.; Yang, H.; Teo, A.S.M.; Amer, L.B.; Sherbaf, F.G.; Tan, C.Q.; Alvarez, J.J.S.; Lu, B.; Lim, J.Q.; Takano, A.; et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat. Genet. 2020, 52, 177–186. [Google Scholar] [CrossRef]
- Block, C.J.; Dyson, G.; Campeanu, I.J.; Watza, D.; Ratnam, M.; Wu, G. A stroma-corrected ZEB1 transcriptional signature is inversely associated with antitumor immune activity in breast cancer. Sci. Rep. 2019, 9, 17807. [Google Scholar] [CrossRef] [Green Version]
- Tulchinsky, E.; Demidov, O.; Kriajevska, M.; Barlev, N.A.; Imyanitov, E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 29–39. [Google Scholar] [CrossRef]
- Pei, L.; He, X.; Li, S.; Sun, R.; Xiang, Q.; Ren, G.; Xiang, T. KRAB zinc-finger protein 382 regulates epithelial-mesenchymal transition and functions as a tumor suppressor, but is silenced by CpG methylation in gastric cancer. Int. J. Oncol. 2018, 53, 961–972. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Fredericks, W.J.; Jensen, D.E.; Speicher, D.W.; Huang, X.P.; Neilson, E.G.; Rauscher, F.J., 3rd. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Develop. 1996, 10, 2067–2078. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Muñoz-Antonia, T.; Cress, W.D. Trim28 contributes to EMT via regulation of E-cadherin and N-cadherin in lung cancer cell lines. PLoS ONE 2014, 9, e101040. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Ishikawa, A.; Shoji, Y.; Imai, T. A genotoxic stress-responsive miRNA, miR-574-3p, delays cell growth by suppressing the enhancer of rudimentary homolog gene in vitro. Int. J. Mol. Sci. 2014, 15, 2971–2990. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wang, H.; He, H.; Niu, H.; Li, Y. Interferon induced transmembrane protein 3 regulates the growth and invasion of human lung adenocarcinoma. Thorac. Cancer 2017, 8, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Huggins, D.N.; LaRue, R.S.; Wang, Y.; Knutson, T.P.; Xu, Y.; Williams, J.W.; Schwertfeger, K.L. Characterizing Macrophage Diversity in Metastasis-Bearing Lungs Reveals a Lipid-Associated Macrophage Subset. Cancer Res. 2021, 81, 5284–5295. [Google Scholar] [CrossRef]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, L.; Cao, L.; Wen, Q. Keratin 17 Promotes Lung Adenocarcinoma Progression by Enhancing Cell Proliferation and Invasion. Med. Sci. Monit. 2018, 24, 4782–4790. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Jones, M.G.; Okimoto, R.A.; Nanjo, S.; Chan, M.M.; Yosef, N.; Bivona, T.G.; Weissman, J.S. Single-cell lineages reveal the rates, routes, and drivers of metastasis in cancer xenografts. Science 2021, 371. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jané-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402. [Google Scholar] [CrossRef]
- Mezheyeuski, A.; Bergsland, C.H.; Backman, M.; Djureinovic, D.; Sjoblom, T.; Bruun, J.; Micke, P. Multispectral imaging for quantitative and compartment-specific immune infiltrates reveals distinct immune profiles that classify lung cancer patients. J. Pathol. 2018, 244, 421–431. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.Y.; Tung, S.R.; Strichartz, G.R.; Håkanson, R. Investigation of the specificity of FK 888 as a tachykinin NK1 receptor antagonist. Br. J. Pharmacol. 1994, 111, 1342–1346. [Google Scholar] [CrossRef] [Green Version]
- Shabbir, M.; Stuart, R. Lestaurtinib, a multitargeted tyrosine kinase inhibitor: From bench to bedside. Expert Opin. Investig. Drugs 2010, 19, 427–436. [Google Scholar] [CrossRef]
- Minturn, J.E.; Evans, A.E.; Villablanca, J.G.; Yanik, G.A.; Park, J.R.; Shusterman, S.; Groshen, S.; Hellriegel, E.T.; Bensen-Kennedy, D.; Matthay, K.K.; et al. Phase I trial of lestaurtinib for children with refractory neuroblastoma: A new approaches to neuroblastoma therapy consortium study. Cancer Chemother. Pharmacol. 2011, 68, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Pareek, S.; Huang, Y.; Nath, A.; Huang, R.S. The success story of drug repurposing in breast cancer. Drug Repurposing Cancer Ther. 2020, 173–190. [Google Scholar] [CrossRef]
- Cancer Stat Facts: Lung and Bronchus Cancer. Available online: https://seer.cancer.gov/statfacts/html/lungb.html (accessed on 26 May 2021).
- Hachey, K.J.; Colson, Y.L. Current innovations in sentinel lymph node mapping for the staging and treatment of resectable lung cancer. Semin. Thorac. Cardiovasc. Surg. 2014, 26, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.Q.; Ding, K.; Strumpf, D.; Weir, B.A.; Meyerson, M.; Pennell, N.; Thomas, R.K.; Naoki, K.; Ladd-Acosta, C.; Liu, N.; et al. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 4417–4424. [Google Scholar] [CrossRef]
- Klug, A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Ann. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef] [Green Version]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschellà, G. Zinc-finger proteins in health and disease. Cell Death Discov. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Wang, Y.; Macfarlan, T.S. The Role of KRAB-ZFPs in Transposable Element Repression and Mammalian Evolution. Trends Genet. TIG 2017, 33, 871–881. [Google Scholar] [CrossRef]
- Addison, J.B.; Koontz, C.; Fugett, J.H.; Creighton, C.J.; Chen, D.; Farrugia, M.K.; Padon, R.R.; Voronkova, M.A.; McLaughlin, S.L.; Livengood, R.H.; et al. KAP1 promotes proliferation and metastatic progression of breast cancer cells. Cancer Res. 2015, 75, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, D.T.; Kurtyka, C.; Rawal, B.; Fulp, W.J.; Haura, E.B.; Cress, W.D. Tripartite motif containing 28 (Trim28) can regulate cell proliferation by bridging HDAC1/E2F interactions. J. Biol. Chem. 2012, 287, 40106–40118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czerwińska, P.; Mazurek, S.; Wiznerowicz, M. The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 2017, 24, 63. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, G.; Wu, J.; Zhou, H.; Zhang, Y.; Miao, Y.; Feng, Y.; Yu, J. Zinc finger protein 668 suppresses non-small cell lung cancer invasion and migration by downregulating Snail and upregulating E-cadherin and zonula occludens-1. Oncol. Lett. 2018, 15, 3806–3813. [Google Scholar] [CrossRef]
- Warzecha, C.C.; Carstens, R.P. Complex changes in alternative pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT). Semin. Cancer Biol. 2012, 22, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Choi, P.S.; Chaffer, C.L.; Labella, K.; Hwang, J.H.; Giacomelli, A.O.; Kim, J.W.; Ilic, N.; Doench, J.G.; Ly, S.H.; et al. An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. eLife 2018, 7. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Buchan, S.L.; Rogel, A.; Al-Shamkhani, A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood 2017, 131, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef]
- Burris, H.A.; Infante, J.R.; Ansell, S.M.; Nemunaitis, J.J.; Weiss, G.R.; Villalobos, V.M.; Sikic, B.I.; Taylor, M.H.; Northfelt, D.W.; Carson, W.E., 3rd; et al. Safety and Activity of Varlilumab, a Novel and First-in-Class Agonist Anti-CD27 Antibody, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 2028–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrends, T.; Bąbała, N.; Xiao, Y.; Yagita, H.; van Eenennaam, H.; Borst, J. CD27 Agonism Plus PD-1 Blockade Recapitulates CD4+ T-cell Help in Therapeutic Anticancer Vaccination. Cancer Res. 2016, 76, 2921–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, S.L.; Fallatah, M.; Thirdborough, S.M.; Taraban, V.Y.; Rogel, A.; Thomas, L.J.; Penfold, C.A.; He, L.Z.; Curran, M.A.; Keler, T.; et al. PD-1 Blockade and CD27 Stimulation Activate Distinct Transcriptional Programs That Synergize for CD8(+) T-Cell-Driven Antitumor Immunity. Clin. Cancer Res. 2018, 24, 2383–2394. [Google Scholar] [CrossRef] [Green Version]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Doebele, R.C.; Kerr, K.M. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol. 2019, 16, 341–355. [Google Scholar] [CrossRef]
- Ye, Q.; Singh, S.; Qian, P.R.; Guo, N.L. Immune-Omics Networks of CD27, PD1, and PDL1 in Non-Small Cell Lung Cancer. Cancers 2021, 13, 4296. [Google Scholar] [CrossRef]
- Andersen, M.E.; Yang, R.S.; French, C.T.; Chubb, L.S.; Dennison, J.E. Molecular circuits, biological switches, and nonlinear dose-response relationships. Environ. Health Perspect. 2002, 110, 971–978. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.H.; Park, A.K.; Dong, S.M.; Ahn, J.H.; Park, W.Y. Global analysis of CpG methylation reveals epigenetic control of the radiosensitivity in lung cancer cell lines. Oncogene 2010, 29, 4725–4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.N.; Tseng, R.C.; Hsu, H.S.; Chen, J.Y.; Tzao, C.; Ho, W.L.; Wang, Y.C. Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer. Clin. Cancer Res. 2007, 13, 832–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.N.; Burgess, J.T.; He, Y.; Gately, K.; Snell, C.; Zhang, S.D.; Hooper, J.D.; Richard, D.J.; O’Byrne, K.J. Expression of CDCA3 Is a Prognostic Biomarker and Potential Therapeutic Target in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1071–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakopoulos, F.D.; Kottorou, A.E.; Kalofonou, M.; Kalofonos, H.P. The Fire Within: NF-κB Involvement in Non-Small Cell Lung Cancer. Cancer Res. 2020, 80, 4025–4036. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Tchou-Wong, K.M.; Rom, W.N. NF-kappaB in lung tumorigenesis. Cancers 2011, 3, 4258–4268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopewell, E.L.; Zhao, W.; Fulp, W.J.; Bronk, C.C.; Lopez, A.S.; Massengill, M.; Antonia, S.; Celis, E.; Haura, E.B.; Enkemann, S.A.; et al. Lung tumor NF-κB signaling promotes T cell-mediated immune surveillance. J. Clin. Investig. 2013, 123, 2509–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Krappmann, D.; Eichten, A.; Heder, A.; Scheidereit, C.; Strauss, M. NF-kappaB function in growth control: Regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell. Biol. 1999, 19, 2690–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.S.; Chen, C.H.; Wang, Y.J.; Pestell, R.G.; Albanese, C.; Chen, R.J.; Chang, M.C.; Jeng, J.H.; Lin, S.Y.; Liang, Y.C.; et al. Tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces cell proliferation in normal human bronchial epithelial cells through NFkappaB activation and cyclin D1 up-regulation. Toxicol. Appl. Pharmacol. 2005, 205, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Allison, D.F.; Baranova, N.N.; Wamsley, J.J.; Katz, A.J.; Bekiranov, S.; Jones, D.R.; Mayo, M.W. NF-κB regulates mesenchymal transition for the induction of non-small cell lung cancer initiating cells. PLoS ONE 2013, 8, e68597. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Lu, S.; Huang, L.; Wang, T.; Wan, Y.; Zhou, C.X.; Zhang, C.; Zhang, Z.; Li, X. The expression of V-ATPase is associated with drug resistance and pathology of non-small-cell lung cancer. Diagn. Pathol. 2013, 8, 145. [Google Scholar] [CrossRef] [Green Version]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starczynowski, D.T.; Lockwood, W.W.; Delehouzee, S.; Chari, R.; Wegrzyn, J.; Fuller, M.; Tsao, M.S.; Lam, S.; Gazdar, A.F.; Lam, W.L.; et al. TRAF6 is an amplified oncogene bridging the RAS and NF-kappaB pathways in human lung cancer. J. Clin. Investig. 2011, 121, 4095–4105. [Google Scholar] [CrossRef] [Green Version]
- Micke, P.; Edlund, K.; Holmberg, L.; Kultima, H.G.; Mansouri, L.; Ekman, S.; Bergqvist, M.; Scheibenflug, L.; Lamberg, K.; Myrdal, G.; et al. Gene copy number aberrations are associated with survival in histologic subgroups of non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1833–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabs, V.; Edlund, K.; Konig, H.; Grinberg, M.; Madjar, K.; Rahnenfuhrer, J.; Ekman, S.; Bergkvist, M.; Holmberg, L.; Ickstadt, K.; et al. Integrative analysis of genome-wide gene copy number changes and gene expression in non-small cell lung cancer. PLoS ONE 2017, 12, e0187246. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Zhan, C.; Zhang, Y.; Ma, J.; Wang, L.; Jiang, W.; Shi, Y.; Wang, Q. Identification of reference genes for qRT-PCR in human lung squamous-cell carcinoma by RNA-Seq. Acta Biochim. Biophys. Sin. 2014, 46, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Walter, R.F.; Werner, R.; Vollbrecht, C.; Hager, T.; Flom, E.; Christoph, D.C.; Schmeller, J.; Schmid, K.W.; Wohlschlaeger, J.; Mairinger, F.D. ACTB, CDKN1B, GAPDH, GRB2, RHOA and SDCBP Were Identified as Reference Genes in Neuroendocrine Lung Cancer via the nCounter Technology. PLoS ONE 2016, 11, e0165181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saviozzi, S.; Cordero, F.; Lo Iacono, M.; Novello, S.; Scagliotti, G.V.; Calogero, R.A. Selection of suitable reference genes for accurate normalization of gene expression profile studies in non-small cell lung cancer. BMC Cancer 2006, 6, 200. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Ding, Y.; Dong, L.; Zhu, L.J.; Jensen, R.V.; Hsiao, L.L. Differential expression patterns of housekeeping genes increase diagnostic and prognostic value in lung cancer. PeerJ 2018, 6, e4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Amgad, M.; Cooper, L.A.D.; Du, Y.; Fu, H.; Ivanov, A.A. High expression of MKK3 is associated with worse clinical outcomes in African American breast cancer patients. J. Translat. Med. 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, D.K.; Lang, J.D.; Rosenthal, H. Prediction Analysis of Cross Classifications; John Wiley & Sons: New York, NY, USA, 1977. [Google Scholar]
- Guo, N.L.; Wan, Y.W.; Bose, S.; Denvir, J.; Kashon, M.L.; Andrew, M.E. A novel network model identified a 13-gene lung cancer prognostic signature. Int. J. Comput. Biol. Drug Des. 2011, 4, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.L.; Wan, Y.W. Pathway-based identification of a smoking associated 6-gene signature predictive of lung cancer risk and survival. Artif. Intell. Med. 2012, 55, 97–105. [Google Scholar] [CrossRef] [Green Version]
- DepMap. DepMap 20Q2 Public. 2020. Available online: https://figshare.com/articles/dataset/DepMap_20Q2_Public/12280541 (accessed on 1 April 2021).
- Aguet, F. TOPMed RNA-Seq Pipeline Harmonization Summary. Available online: https://github.com/broadinstitute/gtex-pipeline/blob/master/TOPMed_RNAseq_pipeline.md (accessed on 24 December 2021).
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019. [Google Scholar] [CrossRef]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addison, J.B.; Voronkova, M.A.; Fugett, J.H.; Lin, C.C.; Linville, N.C.; Trinh, B.; Livengood, R.H.; Smolkin, M.B.; Schaller, M.D.; Ruppert, J.M.; et al. Functional Hierarchy and Cooperation of EMT Master Transcription Factors in Breast Cancer Metastasis. Mol. Cancer Res. 2021, 19, 784–798. [Google Scholar] [CrossRef] [PubMed]
- Team, R. RStudio: Integrated Development Environment for R, 1.4.1106; RStudio, PBC: Boston, MA, USA.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | PRISM | GDSC1 | GDSC2 |
|---|---|---|---|
| Carboplatin | IFITM3, ATP1B4, PRRX1, PRRX1 | ||
| Cisplatin | FUT7 | POF1B, ZNF671, ZBED2, C10orf10 | |
| Docetaxel | ERH, KLHL6 | KLHL6, IFITM3, C10orf10 | SERPING1 |
| Erlotinib | KLHL6, TRIM29, IFITM3, CCR2, TNFSF12-TNFSF13, ATP1B4 | IFITM3, SERPINB5, PRDM1, RGS17, CD80, AKR1C1, TRIM29 | DEFB103A, CSPP1, TRIM29, SERPINB5, AKR1C2, KLHL6, ATP1B4, SERPING1, DSG3 |
| Etoposide | PRDM1, TRIM29 | ATP1B4, SERPING1, ABCA13, ARSE | |
| Gefitinib | DEFB103A, PTAFR, ZEB1, ERH, F13A1 | DEFB103A, KLHL6, TRIM29, RGS17, ATP1B4, PRRX1 | CSPP1, KLHL6, TRIM29, ATP1B4, SERPING1, ARSE |
| Gemcitabine | ARSE, GPX2, RGS17, LAIR1, TRIM22 | SERPING1 | ARSE, C10orf10 |
| Paclitaxel | PTAFR, TRIM29, RAB25, ESRP1 SERPINB5, KLHL6, CD80, SH2D1A | CD27 | C10orf10 |
| Pemetrexed | SH2D1A | ||
| Vinorelbine | FUT7, TNFSF12-TNFSF13, DSG3 |
| Drug | PRISM | GDSC1 | GDSC2 |
|---|---|---|---|
| Carboplatin | TGM5 | ||
| Cisplatin | TRIM29 | CD274, HLA-F | LY75, PRRX1, F13A1, CTSH |
| Docetaxel | SERPINB5 | AKR1C2, SERPINB5 | AKR1C2, AKR1C1, CPNE8 |
| Erlotinib | ARSE, HLA-DMB, TRIM22, LY75, TGM5 | AKR1C1 | POF1B, SERPINB5, F13A1 |
| Etoposide | HLA-F, ABCA13, TGM5 | SERPINB5 | |
| Gefitinib | SLC39A8 | ||
| Gemcitabine | TGM5, HNMT, CTSH, HLA-F, SERPINB5, CDH3, HNMT, ABCA13, AKR1C2, GPX2, AKR1C1 | SERPINB5, AKR1C1 | AKR1C1 |
| Paclitaxel | SERPINB5, CDH3, HLA-F, SLC39A8 | PRRX1, LY75, CTSH, TRIM29 | |
| Pemetrexed | |||
| Vinorelbine | SERPINB5, AKR1C2, AKR1C1, F13A1, HLA-F | CTSH |
| src_set_id | ZNF71 Upregulated Network Genes | ZNF71 Downregulated Network Genes | ||
|---|---|---|---|---|
| Cell Line | Type | Cell Line | Type | |
| BIOCARTA_CTL_PATHWAY | A549 | TRT_SH.CGS | A549 | TRT_SH.CGS |
| HCC515 | TRT_SH.CGS | HCC515 | TRT_SH.CGS | |
| CP_FGFR_INHIBITOR (PD-173074, dovitinib, brivanib, orantinib) | A549 | TRT_CP | A549 | TRT_CP |
| KD_RNA_POLYMERASE_ENZYMES (POLR2A, POLR2C, POLR2D, POLR2E, POLR2F, POLR2I, POLR2K) | A549 | TRT_SH.CGS | HCC515 | TRT_SH.CGS |
| KEGG_GALACTOSE_METABOLISM | HCC515 | TRT_SH.CGS | HCC515 | TRT_SH.CGS |
| PID_CIRCADIAN_PATHWAY | A549 | TRT_SH.CGS | A549 | TRT_SH.CGS |
| PID_INTEGRIN2_PATHWAY | HCC515 | TRT_SH.CGS | A549 | TRT_SH.CGS |
| PID_S1P_S1P2_PATHWAY | HCC515 | TRT_SH.CGS | HCC515 | TRT_SH.CGS |
| REACTOME_CHYLOMICRON_MEDIATED_LIPID_TRANSPORT | HCC515 | TRT_SH.CGS | HCC515 | TRT_SH.CGS |
| REACTOME_JNK_C_JUN_KINASES_PHOSPHORYLATION_AND_ACTIVATION_MEDIATED_BY_ACTIVATED_HUMAN_TAK1 | HCC515 | TRT_SH.CGS | HCC515 | TRT_SH.CGS |
| REACTOME_PURINE_RIBONUCLEOSIDE_MONOPHOSPHATE_BIOSYNTHESIS | A549 | TRT_SH.CGS | A549 | TRT_SH.CGS |
| HCC515 | TRT_SH.CGS | |||
| SULFONYLUREA (glipizide, glibenclamide, gliquidone) | HCC515 | TRT_CP | HCC515 | TRT_CP |
| src_set_id | KRAB Upregulated Network Genes | KRAB Downregulated Network Genes | ||
|---|---|---|---|---|
| Cell Line | Type | Cell Line | Type | |
| BIOCARTA_CTL_PATHWAY | HCC515 | TRT_SH.CGS | HCC515 | TRT_SH.CGS |
| A549 | TRT_SH.CGS | |||
| BIOCARTA_GLYCOLYSIS_PATHWAY | HCC515 | TRT_SH.CGS | A549 | TRT_SH.CGS |
| BIOCARTA_SET_PATHWAY | A549 | TRT_OE | A549 | TRT_OE |
| CP_CCK_RECEPTOR_ANTAGONIST (devazepide, LY-225910, SR-27897) | HCC515 | TRT_CP | HCC515 | TRT_CP |
| CP_FGFR_INHIBITOR (PD-173074, dovitinib, brivanib, orantinib) | A549 | TRT_CP | HCC515 | TRT_CP |
| KD_INTEGRIN_SUBUNITS_BETA (ITGB1, ITGB4, ITGB5) | A549 | TRT_XPR | A549 | TRT_XPR |
| PID_DNA_PK_PATHWAY | A549 | TRT_SH.CGS | A549 | TRT_SH.CGS |
| REACTOME_NFKB_IS_ACTIVATED_AND_SIGNALS_SURVIVAL | A549 | TRT_SH.CGS | HCC515 | TRT_SH.CGS |
| REACTOME_REGULATION_OF_COMPLEMENT_CASCADE | HCC515 | TRT_SH.CGS | HCC515 | TRT_SH.CGS |
| SODIUM/GLUCOSE_COTRANSPORTER_INHIBITOR (maackiain, phloretin) | A549 | TRT_CP | A549 | TRT_CP |
| SULFONYLUREA (glipizide, glibenclamide, gliquidone) | HCC515 | TRT_CP | HCC515 | TRT_CP |
| TGF_BETA_RECEPTOR_INHIBITOR | HCC515 | TRT_CP | HCC515 | TRT_CP |
| src_set_id | KRAB-Less Upregulated Network Genes | KRAB-Less Downregulated Network Genes | ||
|---|---|---|---|---|
| Cell Line | Type | Cell Line | Type | |
| ABL_KINASE_INHIBITOR | A549 | TRT_CP | HCC515 | TRT_CP |
| BIOCARTA_BLYMPHOCYTE_PATHWAY | A549 | TRT_SH.CGS | A549 | TRT_SH.CGS |
| KD_CYCLINS (CCNL1, CCND1, CCNA1, CCNH) | HCC515 | TRT_SH.CGS | A549 | TRT_OE |
| KEGG_CIRCADIAN_RHYTHM_MAMMAL | A549 | TRT_SH.CGS | A549 | TRT_SH.CGS |
| REACTOME_TRAF3_DEPENDENT_IRF_ACTIVATION_PATHWAY | A549 | TRT_SH.CGS | A549 | TRT_SH.CGS |
| src_set_id | Cell_Iname | Pert_Type | Genes |
|---|---|---|---|
| KD_APOLIPOPROTEINS | A549 | TRT_SH.CGS | APOB, APOC2, APOE |
| KD_CYCLINS | A549 | TRT_SH.CGS | CCNL1, CCND1, CCNA1, CCNH |
| HCC515 | TRT_SH.CGS | ||
| KD_LYSINE_ACETYLTRANSFERASES | A549 | TRT_SH.CGS | KAT6B, KAT6A, NCOA3 |
| KD_NON_HOMOLOGOUS_END_JOINING | A549 | TRT_SH.CGS | RAD50, FEN1, XRCC4, XRCC5 |
| KD_V_TYPE_ATPASES | HCC515 | TRT_SH.CGS | ATP6V1A, ATP6V0C, ATP6V0B, ATP6V1F |
| OE_NFKB_ACTIVATION | HCC515 | TRT_SH.CGS | CD40, FADD, LTBR, TNFRSF10A, TNFRSF10B, TNFRSF1A, BCL10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Q.; Falatovich, B.; Singh, S.; Ivanov, A.V.; Eubank, T.D.; Guo, N.L. A Multi-Omics Network of a Seven-Gene Prognostic Signature for Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2022, 23, 219. https://doi.org/10.3390/ijms23010219
Ye Q, Falatovich B, Singh S, Ivanov AV, Eubank TD, Guo NL. A Multi-Omics Network of a Seven-Gene Prognostic Signature for Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2022; 23(1):219. https://doi.org/10.3390/ijms23010219
Chicago/Turabian StyleYe, Qing, Brianne Falatovich, Salvi Singh, Alexey V. Ivanov, Timothy D. Eubank, and Nancy Lan Guo. 2022. "A Multi-Omics Network of a Seven-Gene Prognostic Signature for Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 23, no. 1: 219. https://doi.org/10.3390/ijms23010219