





Abstract
Misfolded protein in the amygdala is a neuropathologic feature of Alzheimer disease and many other neurodegenerative disorders. We examined extracts from human amygdala (snap-frozen at autopsy) to investigate whether novel and as yet uncharacterized misfolded proteins would be detectable. Polypeptides from the detergent-insoluble, urea-soluble protein fractions of amygdala were interrogated using liquid chromatography-electrospray ionization-tandem mass spectrometry. Among the detergent-insoluble proteins identified in amygdala of demented subjects but not controls were Tau, TDP-43, Aβ, α-synuclein, and ApoE. Additional detergent-insoluble proteins from demented subjects in the high-molecular weight portion of SDS gels included NNT, TNIK, PRKDC (DNA-PK, or DNA-PKcs), ferritin light chain (FTL), AIFM1, SYT11, STX1B, EAA1, COL25A1, M4K4, CLH1, SQSTM, SYNJ1, C3, and C4. In follow-up immunohistochemical experiments, NNT, TNIK, PRKDC, AIFM1, and FTL were observed in inclusion body-like structures in cognitively impaired subjects’ amygdalae. Double-label immunofluorescence revealed that FTL and phospho-PRKDC immunoreactivity colocalized partially with TDP-43 and/or Tau inclusion bodies. Western blots showed high-molecular weight “smears”, particularly for NNT and PRKDC. A preliminary genetic association study indicated that rare NNT, TNIK, and PRKDC gene variants had nominally significant association with Alzheimer-type dementia risk. In summary, novel detergent-insoluble proteins, with evidence of proteinaceous deposits, were found in amygdalae of elderly, cognitively impaired subjects.
INTRODUCTION
Dementia is a prevalent and devastating clinical syndrome, usually associated with an underlying neurodegenerative disease (ND), or, commonly, a combination of different diseases (1, 2). Pathologic proteinaceous aggregates in neural tissues are a cardinal feature of NDs (3). For reasons that are currently unknown, specific brain regions are particularly susceptible in the different NDs. Thus, the 2 characteristics that are used to categorize the NDs from a pathologist’s perspective are: (1) the specific polypeptides that are present within proteinaceous deposits in the disease; and (2) the brain regions that are usually most involved during the disease.
At autopsy of persons with dementia, the amygdala typically shows atrophy and cell loss, and, correspondingly, the amygdala harbors pathologic markers referent to multiple different species of misfolded proteins (4–9). More specifically, within the amygdala there is pathologic Tau (neurofibrillary tangles [NFTs]) and Aβ peptide protein (amyloid plaques) in Alzheimer disease (AD), and α-synuclein aggregates (Lewy bodies) in multiple subtypes of Lewy body disease. In addition, TAR-DNA binding protein 43 (TDP-43) pathology is observed commonly in the amygdala as a comorbidity in brains with AD pathology and in cerebral age-related TDP-43 with sclerosis (CARTS) (6–11).
Research on “misfoldingopathies” is evolving rapidly; recent studies on TDP-43 pathology provide an illustrative example. Aggregated TDP-43 protein was first reported as a pathologic marker for frontotemporal lobar degeneration and amyotrophic lateral sclerosis in 2006 (12). Subsequent studies have found that the brains of 20%–50% of aged individuals contain TDP-43 pathology (11, 13–20), which is associated with substantial cognitive impairment (21, 22). From this example of a common and symptomatic brain disease, unknown until recent years, one can suspect the existence of analogous but hitherto unsuspected NDs.
In previous studies in which established pathogenic misfolded proteins were characterized, those polypeptides were isolated from the detergent insoluble protein fraction (12, 23, 24). Here, we apply the term detergent-insoluble pathogenic proteins (DIPPs) to refer to polypeptides that can misfold and provide pathologic markers in the aged human brain. The presence of a given DIPP in an aged person’s brain is increasingly understood to be nonpathognomonic in itself, and comorbid pathologies are more the rule than the exception. Even in early-onset AD cases caused by mutations of the APP gene, there frequently are simultaneous deposits of Tau, α-synuclein, and TDP-43 in addition to Aβ (25–27), often in the amygdala, which suggests the existence of underlying mechanisms that affect multiple polypeptides simultaneously or sequentially.
Many different DIPPs have been identified in neurologically diseased brains, although some of the DIPPs have only been reported in rare conditions. These include gene products of Optineurin (OPTN), Fused in Sarcoma (FUS), and others (28–32). An unanswered question is: are there additional DIPPs (other than Tau, α-synuclein, TDP-43, and Aβ) in aged persons’ brains in association with common, dementia-inducing proteinopathic conditions?
Because the amygdala is a brain area affected by multiple DIPPs in numerous different common NDs, we hypothesized that as yet unidentified DIPPs may be detectable in the amygdala of persons who suffered cognitive deterioration during life. Identifying those novel misfolded pathogenetic proteins could provide both diagnostic and therapeutic targets. One can neither predict a priori which polypeptides those may be, nor depend on animal models to simulate the unique biology of the aged human brain. Instead, we sought an approach anchored in the study of human brain tissue to identify novel candidate DIPPs.
We studied proteins extracted from the amygdalae of individuals who had been followed to autopsy with tissue available at the University of Kentucky AD Center (UK-ADC) biobank. Mass spectrometric proteomic techniques were used to analyze detergent-insoluble protein fractions, allowing identification of novel candidate DIPPs. Follow-up experiments were performed including immunohistochemistry and immunoblotting of extracts from the brains of individuals with cognitive impairment and from controls. Further, a genetic association study identified rare coding variants associated with an AD-type dementia phenotype among the genes that encode the novel candidate DIPPs.
MATERIALS AND METHODS
Biosamples and Fractionation of Amygdala Proteins
Details of UK-ADC research volunteers’ recruitment, inclusion/exclusion criteria, and clinical and pathological assessments have been described previously (1, 33–36). Protocols were approved by the University of Kentucky Institutional Review Board. Research subjects with relatively rare dementia syndromes (e.g., prions, trinucleotide repeat diseases, or frontotemporal lobar degeneration), or any brain tumor were excluded. We also obtained information on agonal events for each subject, and additional criteria for exclusion from the study were an extended interval of premortem hypoxia, any medical ventilator use, brain edema, or large infarct. A convenience sample of cases was selected from among autopsies with a postmortem interval (PMI) of <4 hours. Criteria for constituting an amygdala with disease was having >1 misfolded protein present in the medial temporal lobe, according to neuropathologic examination. Dissected amygdalae were snap-frozen in liquid nitrogen at the time of autopsy and then stored at −80 °C until these experiments were performed.
Fractionation followed the published methodology of Sampathu et al (37), with some modifications. Each amygdala was weighed, then homogenized in 5 mL low salt (LS) buffer (10 mM Tris-HCl, pH7.5, 5 mM EDTA, and 10% (w/v) sucrose) per gram tissue using tissue grinders (VWR #47732-446). The homogenates were centrifuged at 25 000 g, 4 °C for 30 minutes, and the supernatants saved as the LS fractions. The pellets were reextracted with LS buffer, centrifuged as above, and the supernatants were discarded. The pellets were extracted with 5 mL TX buffer (LS buffer supplemented with 1% (v/v) Triton-X-100 and 0.5 M NaCl) per gram original tissue, centrifuged at 180 000 g, 4 °C for 30 minutes, and the supernatants saved as the TX fractions. The pellets were reextracted with TX buffer, centrifuged as above, and the supernatants were discarded. The pellets were extracted with 5 mL myelin flotation buffer (TX buffer containing 30% [w/v] sucrose) per gram original tissue, centrifuged at 180 000 g, 4 °C for 30 minutes, and the supernatants were discarded. The pellets were extracted with 5 mL SARC buffer (LS buffer supplemented with 1% [w/v] N-lauroylsarcosine and 0.5 M NaCl) per gram original tissue, incubated at 22 °C for 2 hours on an end-over-end shaker, centrifuged at 180 000 g, 22 °C for 30 minutes, and the supernatants were saved as the SARC fractions. The pellets were extracted with 0.75 mL urea buffer (7 M urea, 2 M thiourea, 4% (w/v) CHAPS, and 30 mM Tris-HCl, pH8.5) per gram original tissue at 22 °C, centrifuged at 25 000 g, 22 °C for 30 minutes, and the supernatants saved as the urea soluble fractions. The LS, TX, SARC, and myelin flotation buffers were supplemented with protease inhibitor cocktail (Millipore Sigma P8340, 1:300), phenylmethylsulfonyl fluoride ([PMSF], 0.2 mM), N-ethylmaleimide ([NEM], 5 mM), nicotinamide (20 mM), Trichostatin A (1.5 μM), sodium orthovanadate (1 mM), and PhosSTOP phosphatase inhibitor tablets (1 tablet per 10 mL, Millipore Sigma 4906837001). The LS, TX, SARC, and myelin flotation homogenates were supplemented with 1 mM DL-dithiothreitol (DTT) immediately before centrifugation, and the supernatants were saved with SDS-PAGE loading buffer and heated at 94 °C for 5 minutes. The urea (i.e., detergent-insoluble) fractions were saved similarly, but without heating to avoid protein carbamoylation. The protein concentrations of the fractions were determined with Protein Assay Dye Reagent (Cat. #5000006, Bio-Rad, Hercules, CA).
Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry (LC-ESI-MS/MS) and Data Analysis
All mass spectra reported in this study were acquired at the University of Kentucky Proteomics Core Facility (http://www.research.uky.edu/core/proteomics/). Equal total protein amounts of the detergent-insoluble fractions were resolved by denaturing polyacrylamide gel electrophoresis (SDS-PAGE) on 4%–15% gradient gel (Bio-Rad Cat. #4561084), followed by staining with Sypro Ruby protein gel stain (Cat. #S-12000, Molecular Probes, Eugene, OR). Gel pieces were cut followed by dithiothreitol reduction, iodoacetamide alkylation, and in-gel trypsin digestion. The resulting tryptic peptides were extracted, concentrated and subjected to LC-MS/MS analysis as previously described (38, 39). Briefly, LC-MS/MS analysis was performed using an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) coupled with an Eksigent Nanoflex cHiPLC system (Eksigent, Dublin, CA) through a nano-electrospray ionization source. The peptide samples were separated with a reversed phase cHiPLC column (75 μm × 150 mm) at a flow rate of 300 nL/minute. Mobile phase A was water with 0.1% (v/v) formic acid while B was acetonitrile with 0.1% (v/v) formic acid. A 50-minute gradient condition was applied: initial 3% mobile phase B was increased linearly to 40% in 24 minutes and further to 85% and 95% for 5 minutes each before it was decreased to 3% and reequilibrated. LC-MS/MS data were acquired in an information-dependent acquisition mode. Each data collection cycle consisted of 8 scan events: one Orbitrap MS scan (300–1800 m/z) with 60 000 resolution for parent ions followed by MS/MS for fragmentation of the 7 most intense ions using collision induced dissociation (CID).
The LC-MS/MS data referent to gel slices cut from the same lanes were combined and submitted for MS/MS protein identification using the MASCOT algorithm via Proteome Discoverer (version 1.3, Thermo Fisher Scientific), applying a custom database containing human protein sequences from Uniprot. Parameters used in the MASCOT MS/MS ion search were trypsin digestion with maximum of 2 missed cleavages, cysteine carbamidomethylation, and methionine oxidation. Posttranslational modifications were also examined in MASCOT searches, including lysine acetylation, ubiquitylation, as well as serine, threonine, and tyrosine phosphorylation. Mass error tolerance was set to be <10 ppm for MS and 0.8 Da for MS/MS. A decoy database was built and searched to determine the false discovery rates (FDR). Peptides with FDR lower than 0.01 were assigned as high confidence identification. The MASCOT software returns probability-based scores, calculated from the spectra detected of the individual peptides. The scores are reported as −10 × log10(P), where P is the probability that the observed match is a random event. For example, a MASCOT mass spectrometry score of 30 indicates that the probability of a peptide identification being a random event is 0.001.
Immunohistochemistry
The decision as to which specific proteins would be probed with antibodies on fixed brain tissue was made based on availability of a commercial antibody that was suitable for both Western blots and for immunohistochemistry on formalin-fixed, paraffin-embedded tissue, and, known relatively high expression of the protein in the human brain, especially within neurons. Prior to immunohistochemistry, brain tissue was immersion-fixed in 10% neutral-buffered formalin (Cat. #C4320-105, Cardinal Health, Dublin, OH) for 2–4 weeks before paraffin embedding. Immunohistochemical stains were performed as previously described (40) except that formic acid pretreatment was used for all slides. Briefly, sections cut at 8-μm thickness from formalin-fixed-paraffin-embedded tissue blocks were deparaffinized prior to microwave antigen retrieval for 6 minutes (power 8) using citrate buffer (Declere buffer, Cell Marque; Rocklin, CA). See Table 1 for pH used during antigen retrieval for each antibody. The sections were then placed in 100% formic acid (Cat. #A-119 P, Fisher Scientific) for 3 minutes. Sections were blocked in 5% normal goat or rabbit serum in TRIS buffered saline (5% S + TBS) for 1 hour at room temperature, then incubated in primary antibodies diluted in 5% S + TBS, for 22 hours at 4 °C. Secondary antibodies were biotinylated IgG (Cat. #BA-1000, #BA-2000, #BA-4000, and #BA-5000 from Vector Labs, Burlingame, CA) diluted at 1:200 in 5% S + TBS for 1 hour at 19–22 °C. After washing, the Vectastain ABC kit (Vector Labs, Burlingame, CA) was used with Vector Nova Red (Vector Labs), followed by counterstain with hematoxylin, rinsing, dehydration, clearing, and mounting in coverslip media.
Antibodies and Antisera Used in the Current Study to Evaluate Candidate Detergent-Insoluble Pathogenic Proteins
| Antigen | Vendor | Catalog # | Host | AR* | Dilution |
|---|---|---|---|---|---|
| AIFM1 (clone E20) | Abcam | ab32516 | Rabbit | pH 6 | 1:250 |
| AIFM1 | Lifespan Biosciences | LS-C353147 | Rabbit | pH 6 | 1:100 |
| Ferritin light chain | Abcam | ab110017 | Goat | pH 6 | 1:250 |
| Ferritin light chain (clone D-9) | Santa Cruz Biotech | sc74513 | Mouse | pH 6 | 1:100 |
| NNT | Sigma | HPA004829 | Rabbit | pH 6 | 1:200 |
| PRKDC (C-term) | Lifespan Biosciences | LS-B6857 | Rabbit | pH 6 | 1:100 |
| PRKDC(phos) S2056 | Abcam | ab18192 | Rabbit | pH 8 | 1:250 |
| PRKDC(phos) T2609 | Lifespan Biosciences | LS-B409 | Rabbit | pH 8 | 1:200 |
| PRKDC(phos) T2647 | Lifespan Biosciences | LS-C368485 | Rabbit | pH 8 | 1:100 |
| TNIK | Abcam | ab150739 | Rabbit | pH 6 | 1:500 |
| TNIK (clone C-1) | Santa Cruz Biotech | sc377215 | Mouse | pH 6 | 1:100 |
| CLAC-P | Fisher Scientific | PA1-4308 | Rabbit | pH 6 | 1:100 |
| EEA1 (clone G-4) | Santa Cruz Biotech | sc137130 | Mouse | pH 6 | 1:100 |
| MAP4K4 | Abcam | ab155583 | Rabbit | pH 6 | 1:250 |
| Antigen | Vendor | Catalog # | Host | AR* | Dilution |
|---|---|---|---|---|---|
| AIFM1 (clone E20) | Abcam | ab32516 | Rabbit | pH 6 | 1:250 |
| AIFM1 | Lifespan Biosciences | LS-C353147 | Rabbit | pH 6 | 1:100 |
| Ferritin light chain | Abcam | ab110017 | Goat | pH 6 | 1:250 |
| Ferritin light chain (clone D-9) | Santa Cruz Biotech | sc74513 | Mouse | pH 6 | 1:100 |
| NNT | Sigma | HPA004829 | Rabbit | pH 6 | 1:200 |
| PRKDC (C-term) | Lifespan Biosciences | LS-B6857 | Rabbit | pH 6 | 1:100 |
| PRKDC(phos) S2056 | Abcam | ab18192 | Rabbit | pH 8 | 1:250 |
| PRKDC(phos) T2609 | Lifespan Biosciences | LS-B409 | Rabbit | pH 8 | 1:200 |
| PRKDC(phos) T2647 | Lifespan Biosciences | LS-C368485 | Rabbit | pH 8 | 1:100 |
| TNIK | Abcam | ab150739 | Rabbit | pH 6 | 1:500 |
| TNIK (clone C-1) | Santa Cruz Biotech | sc377215 | Mouse | pH 6 | 1:100 |
| CLAC-P | Fisher Scientific | PA1-4308 | Rabbit | pH 6 | 1:100 |
| EEA1 (clone G-4) | Santa Cruz Biotech | sc137130 | Mouse | pH 6 | 1:100 |
| MAP4K4 | Abcam | ab155583 | Rabbit | pH 6 | 1:250 |
AR, antigen retrieval conditions.
Antibodies and Antisera Used in the Current Study to Evaluate Candidate Detergent-Insoluble Pathogenic Proteins
| Antigen | Vendor | Catalog # | Host | AR* | Dilution |
|---|---|---|---|---|---|
| AIFM1 (clone E20) | Abcam | ab32516 | Rabbit | pH 6 | 1:250 |
| AIFM1 | Lifespan Biosciences | LS-C353147 | Rabbit | pH 6 | 1:100 |
| Ferritin light chain | Abcam | ab110017 | Goat | pH 6 | 1:250 |
| Ferritin light chain (clone D-9) | Santa Cruz Biotech | sc74513 | Mouse | pH 6 | 1:100 |
| NNT | Sigma | HPA004829 | Rabbit | pH 6 | 1:200 |
| PRKDC (C-term) | Lifespan Biosciences | LS-B6857 | Rabbit | pH 6 | 1:100 |
| PRKDC(phos) S2056 | Abcam | ab18192 | Rabbit | pH 8 | 1:250 |
| PRKDC(phos) T2609 | Lifespan Biosciences | LS-B409 | Rabbit | pH 8 | 1:200 |
| PRKDC(phos) T2647 | Lifespan Biosciences | LS-C368485 | Rabbit | pH 8 | 1:100 |
| TNIK | Abcam | ab150739 | Rabbit | pH 6 | 1:500 |
| TNIK (clone C-1) | Santa Cruz Biotech | sc377215 | Mouse | pH 6 | 1:100 |
| CLAC-P | Fisher Scientific | PA1-4308 | Rabbit | pH 6 | 1:100 |
| EEA1 (clone G-4) | Santa Cruz Biotech | sc137130 | Mouse | pH 6 | 1:100 |
| MAP4K4 | Abcam | ab155583 | Rabbit | pH 6 | 1:250 |
| Antigen | Vendor | Catalog # | Host | AR* | Dilution |
|---|---|---|---|---|---|
| AIFM1 (clone E20) | Abcam | ab32516 | Rabbit | pH 6 | 1:250 |
| AIFM1 | Lifespan Biosciences | LS-C353147 | Rabbit | pH 6 | 1:100 |
| Ferritin light chain | Abcam | ab110017 | Goat | pH 6 | 1:250 |
| Ferritin light chain (clone D-9) | Santa Cruz Biotech | sc74513 | Mouse | pH 6 | 1:100 |
| NNT | Sigma | HPA004829 | Rabbit | pH 6 | 1:200 |
| PRKDC (C-term) | Lifespan Biosciences | LS-B6857 | Rabbit | pH 6 | 1:100 |
| PRKDC(phos) S2056 | Abcam | ab18192 | Rabbit | pH 8 | 1:250 |
| PRKDC(phos) T2609 | Lifespan Biosciences | LS-B409 | Rabbit | pH 8 | 1:200 |
| PRKDC(phos) T2647 | Lifespan Biosciences | LS-C368485 | Rabbit | pH 8 | 1:100 |
| TNIK | Abcam | ab150739 | Rabbit | pH 6 | 1:500 |
| TNIK (clone C-1) | Santa Cruz Biotech | sc377215 | Mouse | pH 6 | 1:100 |
| CLAC-P | Fisher Scientific | PA1-4308 | Rabbit | pH 6 | 1:100 |
| EEA1 (clone G-4) | Santa Cruz Biotech | sc137130 | Mouse | pH 6 | 1:100 |
| MAP4K4 | Abcam | ab155583 | Rabbit | pH 6 | 1:250 |
AR, antigen retrieval conditions.
Antibodies used to detect previously identified DIPPs were as follows: phospho-Tau (PHF-1, gift from Dr. Peter Davies; 1:500 dilution), phospho-TDP-43 (1D3 clone, EMD Millipore, Billerica, MA; 1:500 dilution), α-synuclein (LB509, gift from Dr. Virginia Lee) and amyloid-β peptide (NAB228, gift from Dr. Eddie Lee; 1:10 000 dilution) which were used to detect NFTs, TDP-43 pathology, α-synucleinopathy, and Aβ plaques, respectively. Otherwise, primary antibodies and antisera used in the current study are depicted in Table 1. According to Lifespan Biosciences Technical Support, referring to the antibodies against PRKDC phosphorylated epitopes (Cat. #LS-B409 and Cat. #LS-C368485): “These products were manufactured at different facilities. They were developed in different labs” (41).
Double-label immunofluorescence experiments were performed using methods previously described (42), with slight modifications. Briefly, sections cut at 8-μm thickness were deparaffinized prior to microwave antigen retrieval for 6 minutes (power 8) using citrate buffer (Declere buffer, Cell Marque). The sections were then placed in 100% formic acid for 3 minutes. Sections were next incubated for 45 seconds at room temperature in a 1× solution of TrueBlack (Cat. #23007, Biotium, Fremont, CA) prepared in 70% ethanol, to reduce auto-fluorescence. Sections were blocked in 5% normal goat serum in TRIS buffered saline (5% S + TBS) for 1 hour at room temperature, then incubated in primary antibodies, diluted in 5% S + TBS, for 22 hours at 4 °C. Secondary antibodies were conjugated to Alexa Fluor probes 594 and 488 (Cat. #A11037 and #A11006, Life Technologies; Carlsbad, CA) diluted 1:200 in 5% S + TBS for 1 hour at room temperature. Slides were coverslipped using Invitrogen ProLong Gold mounting medium with DAPI (Cat. #P36935, Fisher Scientific). Controls were run lacking 1 primary antibody (separately for ferritin light chain [FTL] and Phospho-TDP-43) but with both secondary antibodies (all antibodies at same concentrations, all other steps the same), to ensure that neither autofluorescence nor antibody cross-reaction were leading to false-positive colocalization.
Western Blotting
Proteins were separated by denaturing SDS gel electrophoresis using 4–15% gradient gels (Bio-Rad Cat. #4561086); gels were run at 50V constant. The proteins were transferred to nitrocellulose membrane in transfer buffer containing only 10% methanol. The membranes were blocked and the antibodies were applied in 5% nonfat dry milk in TBST (20 mM Tris-HCl, pH 7.5, 0.8% (w/v) NaCl, 0.1% (v/v) Tween-20) with the exception of the phospho-specific PRKDC antibodies where 5% bovine serum albumin was used instead of milk. Primary antibodies (Table 1) were used that had been applied in immunohistochemistry. The secondary antibodies were HRP-linked antimouse and antirabbit (GE Life Sciences NA931 and NA934, respectively). The blots were developed with the SuperSignal West Pico Chemiluminescent Substrate or the SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Cat. #34078 and #34076), and imaged using a Chemidoc MP Imaging System (Bio-Rad). A separate group of cases was used for replication (see Supplementary Data for clinical and pathologic information on all the cases used for immunoblots).
Genetic Association Study
Whole exome sequence data were obtained from the Alzheimer’s Disease Sequencing Project (ADSP; phs000572.v7.p4) provided by Alzheimer’s Disease Genetic Consortium (ADGC) comprising 18 combined studies, and Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium comprising 6 studies (see refs. (43, 44) and Supplementary Data). Among 10 913 subjects in the unrelated (not family members) sample set of ADSP, those who had AD diagnosis data and were 65 years or older at the last visit or at death were included in this study (n = 10 468). All variants were in Hardy-Weinberg equilibrium (cut-off p value 10−5). Data were analyzed from 5374 AD cases and 5094 controls for associations between the AD phenotype and rare variants in 4 genes: NNT, TNIK, PRKDC, and FTL. Analyzing AIFM1 was not possible since it resides on Chr. X and those data were not available. The p values for the associations between these genes and AD diagnosis were calculated by Fisher exact and the permutation test implemented in PLINK v1.90a (45).
RESULTS
The overall goal of the study was to identify proteins that commonly misfold in the amygdala of elderly persons, some of which we hypothesized would be represented in a small convenience sample. The starting point for the study was the analysis of 7 amygdalae that were snap-frozen in liquid nitrogen during short (<4 hours) PMI autopsies: 4 with documented antemortem cognitive impairment and >1 pathologic marker in amygdala, compared to 3 persons who were documented to be intact cognitively during their last clinical visit, with relatively minor pathologic changes in the amygdala. Information on the biosamples used in LC-ESI-MS/MS experiments is presented in Table 2. The study design and work flow are shown in Figure 1.
Cases Used for Mass Spectrometry and Initial Immunohistochemistry and Western Blots
| Case # | Age Death | Clinical Diagnosis | Braak NFT Stage | Medial temporal lobe pathology | PMI (hours) | ||
|---|---|---|---|---|---|---|---|
| Aβ+? | α-synuclein/Lewy+? | TDP-43+? | |||||
| 1 | 86 | NORMAL | I | N | N | N | 1.8 |
| 2 | 75 | NORMAL | I | N | N | N | 3.5 |
| 3 | 84 | NORMAL | 0 | N | N | N | 2.4 |
| 4 | 87 | AD | III | Y | Y | Y | 1.9 |
| 5 | 84 | MCI | I | N | N | Y | 1.8 |
| 6 | 86 | MCI | II | Y | Y | +/− | 4.0 |
| 7 | 86 | AD | VI | Y | Y | Y | 3.3 |
| Case # | Age Death | Clinical Diagnosis | Braak NFT Stage | Medial temporal lobe pathology | PMI (hours) | ||
|---|---|---|---|---|---|---|---|
| Aβ+? | α-synuclein/Lewy+? | TDP-43+? | |||||
| 1 | 86 | NORMAL | I | N | N | N | 1.8 |
| 2 | 75 | NORMAL | I | N | N | N | 3.5 |
| 3 | 84 | NORMAL | 0 | N | N | N | 2.4 |
| 4 | 87 | AD | III | Y | Y | Y | 1.9 |
| 5 | 84 | MCI | I | N | N | Y | 1.8 |
| 6 | 86 | MCI | II | Y | Y | +/− | 4.0 |
| 7 | 86 | AD | VI | Y | Y | Y | 3.3 |
PMI, postmortem interval.
Cases Used for Mass Spectrometry and Initial Immunohistochemistry and Western Blots
| Case # | Age Death | Clinical Diagnosis | Braak NFT Stage | Medial temporal lobe pathology | PMI (hours) | ||
|---|---|---|---|---|---|---|---|
| Aβ+? | α-synuclein/Lewy+? | TDP-43+? | |||||
| 1 | 86 | NORMAL | I | N | N | N | 1.8 |
| 2 | 75 | NORMAL | I | N | N | N | 3.5 |
| 3 | 84 | NORMAL | 0 | N | N | N | 2.4 |
| 4 | 87 | AD | III | Y | Y | Y | 1.9 |
| 5 | 84 | MCI | I | N | N | Y | 1.8 |
| 6 | 86 | MCI | II | Y | Y | +/− | 4.0 |
| 7 | 86 | AD | VI | Y | Y | Y | 3.3 |
| Case # | Age Death | Clinical Diagnosis | Braak NFT Stage | Medial temporal lobe pathology | PMI (hours) | ||
|---|---|---|---|---|---|---|---|
| Aβ+? | α-synuclein/Lewy+? | TDP-43+? | |||||
| 1 | 86 | NORMAL | I | N | N | N | 1.8 |
| 2 | 75 | NORMAL | I | N | N | N | 3.5 |
| 3 | 84 | NORMAL | 0 | N | N | N | 2.4 |
| 4 | 87 | AD | III | Y | Y | Y | 1.9 |
| 5 | 84 | MCI | I | N | N | Y | 1.8 |
| 6 | 86 | MCI | II | Y | Y | +/− | 4.0 |
| 7 | 86 | AD | VI | Y | Y | Y | 3.3 |
PMI, postmortem interval.
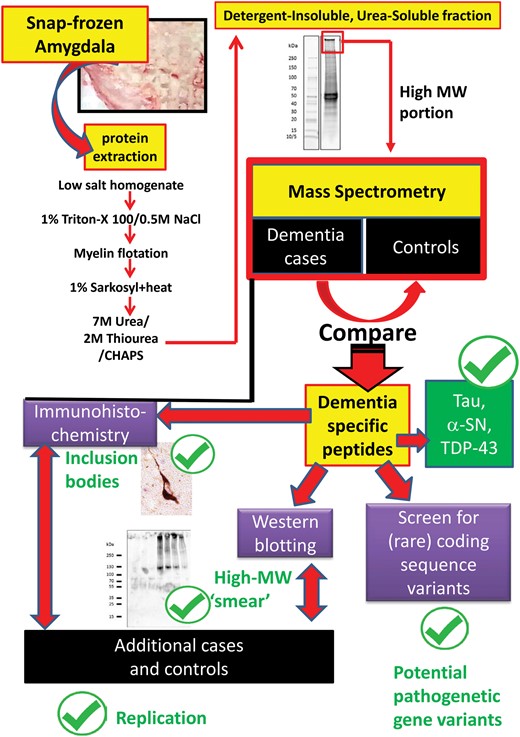
Overview of study design and experiments performed in the current study. With the goal of identifying novel detergent-insoluble pathogenic proteins (DIPPs), experiments began with protein fractionation followed by comparison of high-molecular weight, urea-soluble (detergent-insoluble) proteins from cognitively impaired and control subjects. Optimally, a novel DIPP candidate would be more likely to be present in a preparation that was found to also contain established DIPPs: Tau, α-synuclein, and/or TDP-43. Downstream assessments included Western blots and immunohistochemistry, with separate replication, and with a preliminary genetic screen to evaluate whether rare coding variants could be identified that cosegregated with dementia-inducing disease phenotype.
Proteomic Identification of Candidate DIPPs in Human Amygdala
LC-ESI-MS/MS was used to assess the detergent-insoluble amygdalar proteome within the high-molecular weight (top 9 mm) portion of SDS/denaturing gels, focusing on those polypeptides that were represented in that fraction from 1 or more of the cognitively impaired subjects but not among the control subjects, and with at least 1 sample returning a MASCOT mass spectrometry score of 30. Shown on Table 3 are 19 such polypeptides; a comprehensive list of all 239 proteins identified in the detergent-insoluble fractions is provided in Supplementary Data Table S1 along with more parameters (the number of peptides detected by LC-ESI-MS/MS, the number of unique peptides detected, and the percentage of protein coverage). Four groups of proteins (actually referring to peptide fragments rather than entire proteins) are highlighted on Table 3:
A Subset of Proteins Identified With MASCOT Mass Spectrometry Score >30 in the Detergent-Insoluble, Urea-Soluble, High-Molecular Weight Fraction From Cognitively Impaired Subjects (Cases 4–7 from Table 2) but not Controls
| Accession | Protein ID | MASCOT Mass Spectrometry Score | Established DIPP? | Studied with IHC? | Studied and Showed Inclusions? | ||||
|---|---|---|---|---|---|---|---|---|---|
| Group #* | Case 4 | Case 5 | Case 6 | Case 7 | |||||
| 1 | P02649 | APOE | 231.9 | 233.0 | Y | ||||
| P10636 | TAU | 158.5 | 24.9 | 453.3 | Y | ||||
| P37840 | SNCA | 129.6 | Y | ||||||
| Q13148 | TDP-43 | 85.9 | 71.6 | Y | |||||
| 2 | Q13423 | NNT | 125.0 | 171.7 | 73.3 | 21.7 | Y | Y | |
| P78527 | PRKDC | 42.4 | 176.2 | 32.5 | Y | Y | |||
| O95831 | AIFM1 | 38.8 | 23.4 | Y | Y | ||||
| Q9UKE5 | TNIK | 20.6 | 60.6 | Y | Y | ||||
| P02792 | FTL | 66.3 | 22.8 | Y | Y | ||||
| 3 | Q9BT88 | SYT11 | 50.3 | Y | |||||
| Q9BXS0 | COPA1 | 35.6 | 35.3 | Y | |||||
| O95819 | M4K4 | 20.6 | 60.6 | Y | |||||
| P43003 | EAA1 | 82.9 | 111.4 | Y | |||||
| 4 | P61266 | STX1B | 51.7 | ||||||
| P01024 | C3 | 102.6 | 143.9 | ||||||
| P0C0L4 | C4 | 101.5 | 179.0 | ||||||
| O43426 | SYNJ1 | 78.2 | |||||||
| Q00610 | CLH1 | 58.5 | 29.9 | ||||||
| Q13501 | SQSTM | 38.9 | |||||||
| Accession | Protein ID | MASCOT Mass Spectrometry Score | Established DIPP? | Studied with IHC? | Studied and Showed Inclusions? | ||||
|---|---|---|---|---|---|---|---|---|---|
| Group #* | Case 4 | Case 5 | Case 6 | Case 7 | |||||
| 1 | P02649 | APOE | 231.9 | 233.0 | Y | ||||
| P10636 | TAU | 158.5 | 24.9 | 453.3 | Y | ||||
| P37840 | SNCA | 129.6 | Y | ||||||
| Q13148 | TDP-43 | 85.9 | 71.6 | Y | |||||
| 2 | Q13423 | NNT | 125.0 | 171.7 | 73.3 | 21.7 | Y | Y | |
| P78527 | PRKDC | 42.4 | 176.2 | 32.5 | Y | Y | |||
| O95831 | AIFM1 | 38.8 | 23.4 | Y | Y | ||||
| Q9UKE5 | TNIK | 20.6 | 60.6 | Y | Y | ||||
| P02792 | FTL | 66.3 | 22.8 | Y | Y | ||||
| 3 | Q9BT88 | SYT11 | 50.3 | Y | |||||
| Q9BXS0 | COPA1 | 35.6 | 35.3 | Y | |||||
| O95819 | M4K4 | 20.6 | 60.6 | Y | |||||
| P43003 | EAA1 | 82.9 | 111.4 | Y | |||||
| 4 | P61266 | STX1B | 51.7 | ||||||
| P01024 | C3 | 102.6 | 143.9 | ||||||
| P0C0L4 | C4 | 101.5 | 179.0 | ||||||
| O43426 | SYNJ1 | 78.2 | |||||||
| Q00610 | CLH1 | 58.5 | 29.9 | ||||||
| Q13501 | SQSTM | 38.9 | |||||||
DIPP, detergent-insoluble, pathogenic protein; IHC, immunohistochemistry.
For Group #, see Results.
A Subset of Proteins Identified With MASCOT Mass Spectrometry Score >30 in the Detergent-Insoluble, Urea-Soluble, High-Molecular Weight Fraction From Cognitively Impaired Subjects (Cases 4–7 from Table 2) but not Controls
| Accession | Protein ID | MASCOT Mass Spectrometry Score | Established DIPP? | Studied with IHC? | Studied and Showed Inclusions? | ||||
|---|---|---|---|---|---|---|---|---|---|
| Group #* | Case 4 | Case 5 | Case 6 | Case 7 | |||||
| 1 | P02649 | APOE | 231.9 | 233.0 | Y | ||||
| P10636 | TAU | 158.5 | 24.9 | 453.3 | Y | ||||
| P37840 | SNCA | 129.6 | Y | ||||||
| Q13148 | TDP-43 | 85.9 | 71.6 | Y | |||||
| 2 | Q13423 | NNT | 125.0 | 171.7 | 73.3 | 21.7 | Y | Y | |
| P78527 | PRKDC | 42.4 | 176.2 | 32.5 | Y | Y | |||
| O95831 | AIFM1 | 38.8 | 23.4 | Y | Y | ||||
| Q9UKE5 | TNIK | 20.6 | 60.6 | Y | Y | ||||
| P02792 | FTL | 66.3 | 22.8 | Y | Y | ||||
| 3 | Q9BT88 | SYT11 | 50.3 | Y | |||||
| Q9BXS0 | COPA1 | 35.6 | 35.3 | Y | |||||
| O95819 | M4K4 | 20.6 | 60.6 | Y | |||||
| P43003 | EAA1 | 82.9 | 111.4 | Y | |||||
| 4 | P61266 | STX1B | 51.7 | ||||||
| P01024 | C3 | 102.6 | 143.9 | ||||||
| P0C0L4 | C4 | 101.5 | 179.0 | ||||||
| O43426 | SYNJ1 | 78.2 | |||||||
| Q00610 | CLH1 | 58.5 | 29.9 | ||||||
| Q13501 | SQSTM | 38.9 | |||||||
| Accession | Protein ID | MASCOT Mass Spectrometry Score | Established DIPP? | Studied with IHC? | Studied and Showed Inclusions? | ||||
|---|---|---|---|---|---|---|---|---|---|
| Group #* | Case 4 | Case 5 | Case 6 | Case 7 | |||||
| 1 | P02649 | APOE | 231.9 | 233.0 | Y | ||||
| P10636 | TAU | 158.5 | 24.9 | 453.3 | Y | ||||
| P37840 | SNCA | 129.6 | Y | ||||||
| Q13148 | TDP-43 | 85.9 | 71.6 | Y | |||||
| 2 | Q13423 | NNT | 125.0 | 171.7 | 73.3 | 21.7 | Y | Y | |
| P78527 | PRKDC | 42.4 | 176.2 | 32.5 | Y | Y | |||
| O95831 | AIFM1 | 38.8 | 23.4 | Y | Y | ||||
| Q9UKE5 | TNIK | 20.6 | 60.6 | Y | Y | ||||
| P02792 | FTL | 66.3 | 22.8 | Y | Y | ||||
| 3 | Q9BT88 | SYT11 | 50.3 | Y | |||||
| Q9BXS0 | COPA1 | 35.6 | 35.3 | Y | |||||
| O95819 | M4K4 | 20.6 | 60.6 | Y | |||||
| P43003 | EAA1 | 82.9 | 111.4 | Y | |||||
| 4 | P61266 | STX1B | 51.7 | ||||||
| P01024 | C3 | 102.6 | 143.9 | ||||||
| P0C0L4 | C4 | 101.5 | 179.0 | ||||||
| O43426 | SYNJ1 | 78.2 | |||||||
| Q00610 | CLH1 | 58.5 | 29.9 | ||||||
| Q13501 | SQSTM | 38.9 | |||||||
DIPP, detergent-insoluble, pathogenic protein; IHC, immunohistochemistry.
For Group #, see Results.
Group 1: Established DIPPs that were present in the high-molecular weight detergent-insoluble proteome (Aβ peptide was identified by LC-ESI-MS/MS in the high molecular weight SDS-PAGE gel slice, and lower gel slices, from Case 7, but not in a replication run, so is not shown in Table 3); Group 2: Novel candidate DIPPs for which immunohistochemical staining was performed in the current study, and the respective antibodies detected possible inclusion bodies; Group 3: Novel candidate DIPPs for which immunohistochemical staining was performed and we found no evidence of inclusion bodies in a small preliminary set of brain sections; and Group 4: Novel candidate DIPPs that we did not perform any immunohistochemistry for. (A small number of these are listed in Table 3, whereas all are presented in Supplementary Data Table S1).
Note that none of the Group 1–4 polypeptides were identified in analyses of detergent-insoluble fractions of control brains (Table 2, Cases 1–3). A longer list of polypeptides detected by LC-ESI-MS/MS from urea-soluble fractions of both cases and controls is presented in Supplementary Data Table S1. For the novel candidate DIPPs for which the cognate antibodies highlighted inclusion body-like profiles on immunohistochemistry (Group 2), we assessed the mass spectra for evidence of posttranslational modifications. Extensive posttranslational modifications (particularly phosphorylation) were noted for PRKDC, and there also was evidence of more focal post-translational modifications of NNT, TNIK, and AIFM1, but not FTL; these data are provided in detail in Supplementary Data Table S2.
Immunohistochemistry
Given the small sample size of control subjects analyzed in LC-ESI-MS/MS studies, it was considered likely that at least some of the candidate DIPPs would be false-positives. To follow up the proteomics work, immunohistochemical experiments were performed on a subset of the polypeptides identified as being in the detergent-insoluble fraction from cognitively impaired subjects’ amygdalae. Antibodies against 9 different proteins were used on human amygdala sections from 3 cases with dementia. Whenever possible, we obtained at least a second commercially available antibody for the proteins that had suggestive results in the preliminary analyses, and the staining for these are discussed in greater detail below. Negative for evidence of the proteins being represented in inclusion body-like immunolabeled structures in our hands were CLAC-P/COL25A1, EAA1/SLC1A3, SYT11, and M4K4 (Group 3 proteins, Table 3; data not shown on the immunohistochemical results that were negative).
In the initial immunohistochemistry studies, the following 5 proteins showed immunohistochemical staining that resembled pathologic inclusion bodies according to a neuropathologist (author P.T.N.): PRKDC, FTL, TNIK, NNT, and AIFM1 (Group 2 polypeptides). Antibodies that recognize these 5 proteins were then used to probe fixed amygdala sections from 12 different brains. These cases encompassed a broad spectrum of disease severity, from intact cognition to severe dementia, with a range of comorbid pathologies. The digested results, which include semiquantitative assessment of the amount of immunohistochemical staining for each protein on a section of amygdala from each case, are shown in Table 4. For this table, the following antibodies were used: anti-NNT (Millipore Sigma, HPA004829), anti-PRKDC (Abcam anti-P2056, ab18192), anti-FTL (Santa Cruz, sc74513), anti-TNIK (Santa Cruz, sc377215), and anti-AIFM1 (LSBio, LS-C353147). The immunohistochemical results are discussed in greater detail below for each of the candidate DIPPs.
Immunohistochemical Staining for AIFM1, FTL, NNT, PRKDC, and TNIK in Amygdala Sections From a Convenience Sample of 12 Test Cases From the University of Kentucky AD Center Biobank
| Age | Sex | CDR (G) | Clin Dx | Antibody Staining Inclusion Body-Like Structures* | Braak NFT Stage | Medial Temporal Lobe Neuropathology | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PRKDC | FTL | AIFM1 | TNIK | NNT | Aβ | α-SN | TDP-43 | |||||
| 80 | F | 0 | NL | 0 | 1 | 0 | 0 | 0 | I | N | N | 0 |
| 77 | M | 0 | NL | 1 | 1 | 0 | 0 | 0 | I | N | N | +/− |
| 95 | F | 0 | NL | 1 | 0 | 0 | +/− | 0 | I | N | N | 0 |
| 84 | F | 0.5 | MCI | 2 | 1 | 2 | 2 | 0 | I | N | N | 2 |
| 91 | M | 0.5 | MCI | 1 | 1 | 1 | +/− | 0 | II | N | N | 0 |
| 91 | F | 0.5 | MCI | 2 | 0 | +/− | 0 | 1 | III | Y | N | 0 |
| 97 | M | 1 | PAD | +/− | 1 | 0 | +/− | 0 | II | N | N | 0 |
| 87 | F | 3 | PAD | 1 | 0 | +/− | 0 | 1 | II | Y | Y | 0 |
| 88 | F | 1 | PAD | 1 | 0 | +/− | +/− | 1 | II | N | N | 2 |
| 87 | F | 2 | PAD | 2 | 2 | 2 | 2 | 2 | III | Y | Y | 2 |
| 89 | F | 3 | PAD | 1 | 0 | +/− | +/− | 1 | IV | Y | N | 2 |
| 87 | F | 3 | PAD | 2 | 1 | 2 | 2 | 1 | VI | Y | Y | 2 |
| Age | Sex | CDR (G) | Clin Dx | Antibody Staining Inclusion Body-Like Structures* | Braak NFT Stage | Medial Temporal Lobe Neuropathology | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PRKDC | FTL | AIFM1 | TNIK | NNT | Aβ | α-SN | TDP-43 | |||||
| 80 | F | 0 | NL | 0 | 1 | 0 | 0 | 0 | I | N | N | 0 |
| 77 | M | 0 | NL | 1 | 1 | 0 | 0 | 0 | I | N | N | +/− |
| 95 | F | 0 | NL | 1 | 0 | 0 | +/− | 0 | I | N | N | 0 |
| 84 | F | 0.5 | MCI | 2 | 1 | 2 | 2 | 0 | I | N | N | 2 |
| 91 | M | 0.5 | MCI | 1 | 1 | 1 | +/− | 0 | II | N | N | 0 |
| 91 | F | 0.5 | MCI | 2 | 0 | +/− | 0 | 1 | III | Y | N | 0 |
| 97 | M | 1 | PAD | +/− | 1 | 0 | +/− | 0 | II | N | N | 0 |
| 87 | F | 3 | PAD | 1 | 0 | +/− | 0 | 1 | II | Y | Y | 0 |
| 88 | F | 1 | PAD | 1 | 0 | +/− | +/− | 1 | II | N | N | 2 |
| 87 | F | 2 | PAD | 2 | 2 | 2 | 2 | 2 | III | Y | Y | 2 |
| 89 | F | 3 | PAD | 1 | 0 | +/− | +/− | 1 | IV | Y | N | 2 |
| 87 | F | 3 | PAD | 2 | 1 | 2 | 2 | 1 | VI | Y | Y | 2 |
CDR(G), global clinical dementia rating (0–3 scale); Clin Dx, final clinical diagnosis; PAD, final clinical diagnosis was probable Alzheimer disease; MCI, mild cognitive impairment; NL, cognitively intact.
Semiquantitative scale: 0 = no inclusions seen; +/− = equivocal; 1 = scattered immunoreactive inclusions, most or almost all <2 per high-power microscope field; 2 = common >2 inclusions per high-power field.
Immunohistochemical Staining for AIFM1, FTL, NNT, PRKDC, and TNIK in Amygdala Sections From a Convenience Sample of 12 Test Cases From the University of Kentucky AD Center Biobank
| Age | Sex | CDR (G) | Clin Dx | Antibody Staining Inclusion Body-Like Structures* | Braak NFT Stage | Medial Temporal Lobe Neuropathology | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PRKDC | FTL | AIFM1 | TNIK | NNT | Aβ | α-SN | TDP-43 | |||||
| 80 | F | 0 | NL | 0 | 1 | 0 | 0 | 0 | I | N | N | 0 |
| 77 | M | 0 | NL | 1 | 1 | 0 | 0 | 0 | I | N | N | +/− |
| 95 | F | 0 | NL | 1 | 0 | 0 | +/− | 0 | I | N | N | 0 |
| 84 | F | 0.5 | MCI | 2 | 1 | 2 | 2 | 0 | I | N | N | 2 |
| 91 | M | 0.5 | MCI | 1 | 1 | 1 | +/− | 0 | II | N | N | 0 |
| 91 | F | 0.5 | MCI | 2 | 0 | +/− | 0 | 1 | III | Y | N | 0 |
| 97 | M | 1 | PAD | +/− | 1 | 0 | +/− | 0 | II | N | N | 0 |
| 87 | F | 3 | PAD | 1 | 0 | +/− | 0 | 1 | II | Y | Y | 0 |
| 88 | F | 1 | PAD | 1 | 0 | +/− | +/− | 1 | II | N | N | 2 |
| 87 | F | 2 | PAD | 2 | 2 | 2 | 2 | 2 | III | Y | Y | 2 |
| 89 | F | 3 | PAD | 1 | 0 | +/− | +/− | 1 | IV | Y | N | 2 |
| 87 | F | 3 | PAD | 2 | 1 | 2 | 2 | 1 | VI | Y | Y | 2 |
| Age | Sex | CDR (G) | Clin Dx | Antibody Staining Inclusion Body-Like Structures* | Braak NFT Stage | Medial Temporal Lobe Neuropathology | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PRKDC | FTL | AIFM1 | TNIK | NNT | Aβ | α-SN | TDP-43 | |||||
| 80 | F | 0 | NL | 0 | 1 | 0 | 0 | 0 | I | N | N | 0 |
| 77 | M | 0 | NL | 1 | 1 | 0 | 0 | 0 | I | N | N | +/− |
| 95 | F | 0 | NL | 1 | 0 | 0 | +/− | 0 | I | N | N | 0 |
| 84 | F | 0.5 | MCI | 2 | 1 | 2 | 2 | 0 | I | N | N | 2 |
| 91 | M | 0.5 | MCI | 1 | 1 | 1 | +/− | 0 | II | N | N | 0 |
| 91 | F | 0.5 | MCI | 2 | 0 | +/− | 0 | 1 | III | Y | N | 0 |
| 97 | M | 1 | PAD | +/− | 1 | 0 | +/− | 0 | II | N | N | 0 |
| 87 | F | 3 | PAD | 1 | 0 | +/− | 0 | 1 | II | Y | Y | 0 |
| 88 | F | 1 | PAD | 1 | 0 | +/− | +/− | 1 | II | N | N | 2 |
| 87 | F | 2 | PAD | 2 | 2 | 2 | 2 | 2 | III | Y | Y | 2 |
| 89 | F | 3 | PAD | 1 | 0 | +/− | +/− | 1 | IV | Y | N | 2 |
| 87 | F | 3 | PAD | 2 | 1 | 2 | 2 | 1 | VI | Y | Y | 2 |
CDR(G), global clinical dementia rating (0–3 scale); Clin Dx, final clinical diagnosis; PAD, final clinical diagnosis was probable Alzheimer disease; MCI, mild cognitive impairment; NL, cognitively intact.
Semiquantitative scale: 0 = no inclusions seen; +/− = equivocal; 1 = scattered immunoreactive inclusions, most or almost all <2 per high-power microscope field; 2 = common >2 inclusions per high-power field.
PRKDC: Four different polyclonal antisera against PRKDC were purchased from commercial vendors. One of these antisera recognized a nonphosphorylated epitope. Each of the other 3 antisera recognizes an epitope that includes a phosphorylated residue: Ser2056, Thr2609, or Thr2647, respectively, all of which were implicated in activation of the PRKDC protein (46). These phosphorylation-specific antisera were included since we identified many phosphorylated residues in the LC-MS/MS data analysis (Supplementary Data Table S2). The antiserum raised against nonphosphorylated PRKDC stained cells indistinctly, with high background (Fig. 2). All 3 of the antisera against phosphorylated residues stained similar patterns, but were not identical to each other (Fig. 2): each of these antisera stained what appeared to be clear-cut pathologic inclusion bodies in the brains of demented subjects, as well as distinct neuropil thread-like structures. All 3 antisera also intensely stained roundish (mostly ∼1–3 μm in diameter) structures within neurons—far more in cognitively impaired than control subjects. Double-label immunofluorescence showed that phospho-PRKDC-immunoreactive cells could be double-labeled using antibodies that stain phospho-TDP-43 or phospho-Tau (Fig. 3). For the antiserum against phospho-T2609 PRKDC, the single-label results looked so histomorphologically similar to Tau pathology that this antiserum is quite possibly cross-reacting with phospho-Tau. However, double-label immunofluorescence showed that some phospho-T2609 PRKDC-immunoreactive structures were not immunoreactive for phospho-Tau, and vice versa (Fig. 3).
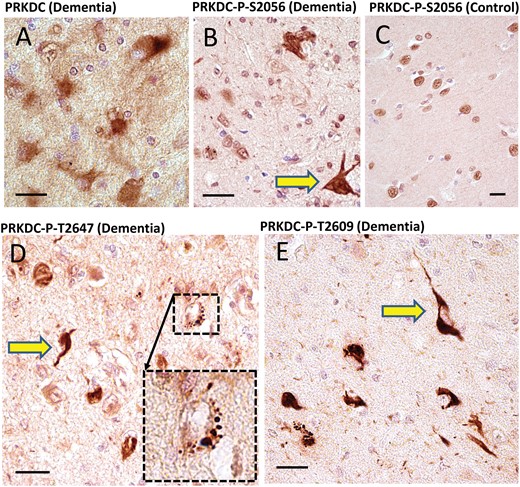
Immunohistochemistry for PRKDC in human brains using 4 separate antisera. Among all the candidate novel DIPPs, the strongest immunohistochemical staining was found using antisera against PRKDC. The antiserum raised against nonphosphorylated PRKDC provided relatively indistinct staining of cellular features, and high background (A). By contrast, all 3 antisera raised against phosphorylated epitopes within PRKDC showed immunostaining of what appeared to be clear-cut inclusion bodies: anti-PRKDC-P (S2056) (B, C); anti-PRKDC-P (T2647) (D); and anti-PRKDC-P (T2609) (E). These results are representative of a tendency to stain inclusion bodies (yellow arrows) in dementia cases. By contrast, nondemented cases showed no inclusion body-like structures, and generally immunostained nuclear profiles, as expected (C). In addition to immunostaining inclusion body-like structures, the anti-PRKDC-P antisera also highlighted roundish cytoplasmic structures in cognitively impaired subjects, such as the one in the inset in panel D. Note also the presence of neuropil thread-like staining (E). Scale bars: A–E = 20 μm.
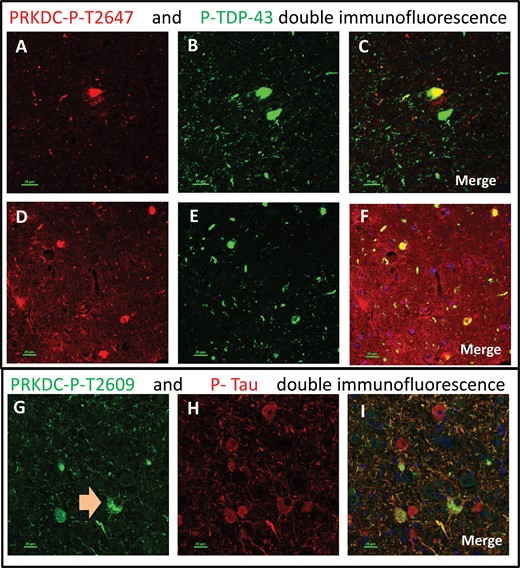
Double-label immunofluorescence: phosphorylated PRKDC. Different antisera (originating from separate laboratories) were used that recognized phosphorylated PRKDC (P-PRKDC). Although the staining results for these antisera were not identical, 3 types of structures were consistently labeled using all 3 antisera: tangle-like intraneuronal inclusions; thread-like extracellular structures; and ∼1 μm radius spherical structures within neurons. Panels A–C and D–F depict the same microscopic field stained for Thr-2647-P-PRKDC (red; A, D) and P-TDP-43 (green; B, E). The merged photomicrographs (C, F) show colocalization in yellow. Note that some but not all cells show colocalized immunoreactivity for both antigens. Panels G–I depict the same microscopic field stained for Thr-2609-P-PRKDC (green) and P-Tau (red). Some but not all cells show colocalized immunoreactivity for both antigens. Note the double-labeled cell with both P-PRKDC spherical structures and P-Tau immunoreactivity (G, arrow). Scale bars: 20 μm.
FTL: One polyclonal antiserum and one monoclonal antibody against FTL were used (Table 1). Both showed similar results. Consistent with the prior published work using laser-captured microdissection and RNA-Seq (47), the FTL immunoreactivity signal was strongest in cells with histomorphologic features of microglia and astrocytes. In control tissue, these stained immunoreactive structures were relatively thin and wispy. In dementia cases, the immunoreactive structures were dense, and included many cells resembling reactive astrocytes and/or microglia, and also some FTL-immunoreactive structures that had histomorphologic features of inclusion bodies (Fig. 4). Some FTL immunoreactivity was present in cells with neuronal features. Double-label immunofluorescence experiments (Fig. 5) showed that FTL immunoreactive structures—some within large glial-like cells, and some that were not clearly associated with specific cellular structures in the plane of section—were colocalized with phospho-TDP-43 immunoreactivity. As immunofluorescence controls, staining reactions were performed lacking 1 or other primary antibody, but with both fluorescence-labeled secondary antibodies. These studies indicated that the colocalization experiments were not due to cross-reaction or to autofluorescence (Supplementary Data Fig. S1).
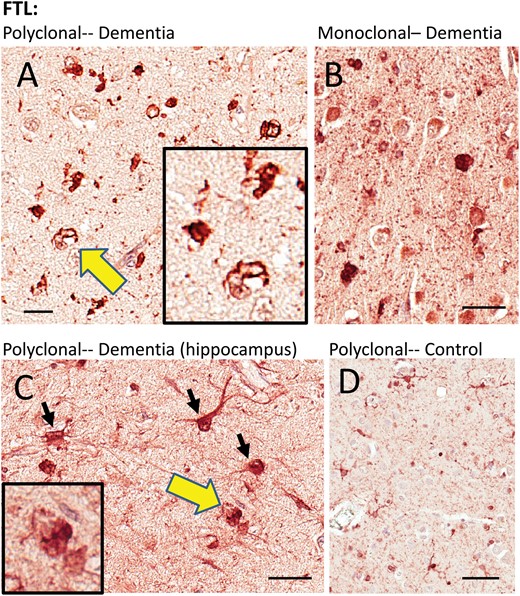
Immunohistochemistry for FTL in human brains. Two different FTL antibodies were used and they showed similar results. A goat polyclonal antiserum (A, C, D) highlighted immunostained structures with astrocyte and microglial features, as well as what looked like intracellular inclusions. A mouse monoclonal anti-FTL antibody (B) showed similar changes as the polyclonal antiserum. Areas indicated with the yellow arrows are shown at higher magnification (insets), depicting FTL immunostained structures with histomorphologic features of inclusion bodies. The cell types stained by the FTL antibodies, even in diseased brains, appeared mostly nonneuronal. Hippocampus from 1 brain with HS-Aging/CARTS was stained (92-year-old at death; (C) to allow better discrimination of cell types, and showed both glial and neuronal (inset) immunostaining. In brains of nondemented subjects (D), FTL antibodies showed relatively sparse staining of cells with quiescent-type microglial and astrocyte morphology. Scale bars: A = 15 μm; B, C = 30 μm; D = 60 μm.
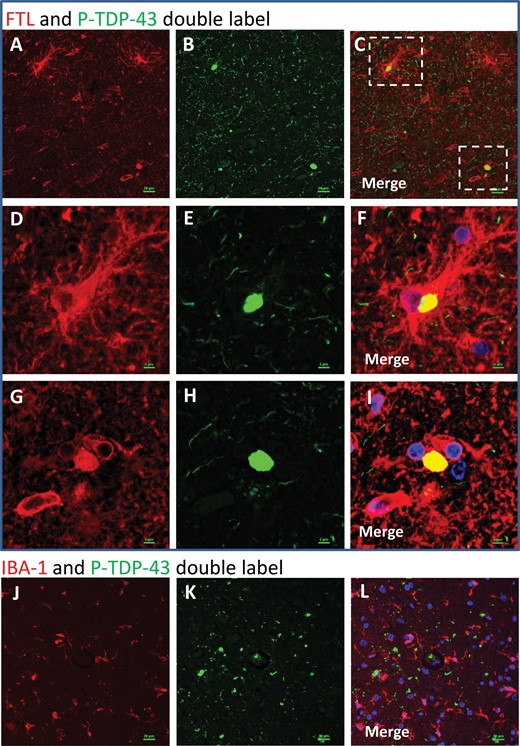
Double-label immunofluorescence: FTL and P-TDP-43. In double-label immunofluorescence experiments, FTL (red) and P-TDP-43 (green) signal overlapped (A–I). Note that the overlap between FTL and P-TDP-43 is only partial – some FTL and some P-TDP-43 signals are not colocalized. Panels D–F and G–I are higher-powered photomicrographs related to the areas of panels A–C that are boxed in panel C. Panels D–F depict a P-TDP-43 immunoreactive inclusion within a large and ramified FTL-immunopositive cell. Panels G–-I depict a P-TDP-43-immunoreactive structure that appears not to be within a cell in the plane of section, but is colocalized with FTL immunoreactivity. As expected from brightfield immunohistochemical experiments (Fig. 4), some noncolocalizing FTL label is present in cells with a microglial morphology. By contrast, when IBA-1 (a different microglial marker, labeled red) and P-TDP-43 (green) antibodies are used (J–L), there does not appear to be any colocalization. Scale bars: A–C, J–L = 15 μm; D–I = 5 μm.
AIFM1: One polyclonal antiserum and one monoclonal antibody against AIFM1 were used (Table 1). Both showed similar results (Fig. 6). No brain section showed extensive widespread AIFM1-immunoreactive inclusion bodies but several of the cases analyzed showed scattered immunoreactive structures that looked like inclusion bodies. The control brains were quite negative for specific immunoreactivity. Notably, both of the AIFM1 antibodies stained corpora amylacea (not shown), which could make filtering background staining a challenge in some of the peripheral portions of amygdala sections.
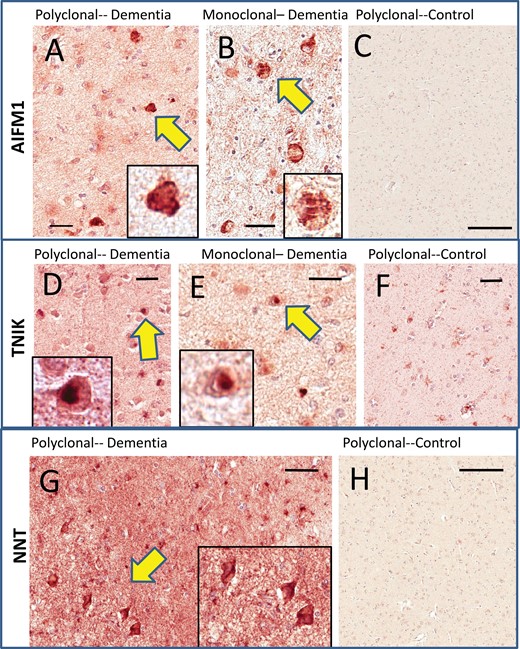
Immunohistochemistry for TNIK, AIFM1, and NNT in human amygdalae. Two different AIFM1 antibodies (A, B) were used and they showed similar results with intracellular-immunoreactive structures that resembled inclusion bodies (see arrows and insets at higher magnification) in cases with cognitive impairment. Immunoreactivity was very sparse in cognitively intact persons (C). Two different TNIK antibodies (D, E) were used and they showed similar results. These resembled Lewy bodies in terms of histopathologic appearance (see insets), and tended to be present in cases with previously diagnosed Lewy body pathology. Control cases showed relatively sparse staining (F). The NNT antiserum showed scattered immunoreactive cells that were more equivocal in terms of inclusion body-type histomorphology (G). However, the background staining for the cases with cognitive impairments was considerably higher than for the cases with intact cognition, which showed only light immunoreactivity (H). Scale bars: A = 35 μm; B = 20 μm; C = 500 μm; D = 30 μm; E = 25 μm; F = 60 μm; G = 50 μm; H = 500 μm.
TNIK: One polyclonal antiserum and one monoclonal antibody against TNIK were used (Table 1). Both showed similar results (Fig. 6). Several of the cases analyzed showed immunoreactive structures that resembled inclusion bodies. These structures had the histomorphologic appearance of Lewy bodies (round intracellular dense staining material) and were present preferentially in cases with known Lewy body disease (Tables 2 and 4). The control brains had relatively low TNIK immunoreactivity, with small immunostained cells showing histomorphologic features of small astrocytes and/or microglial elements with delicate branches. The monoclonal TNIK antibody also stained corpora amylacea (not shown), so the polyclonal antiserum served as a preferable reagent in this context.
NNT: Only a single commercially available antiserum was identified that could recognize NNT for immunohistochemical and Western blotting applications. In terms of labeling inclusion bodies on immunohistochemistry, NNT was the most equivocal of the 5 proteins that were followed up as potential DIPPs (Fig. 6). Some of the NNT-immunoreactive cells were clearly neurons. More remarkable than the relatively scattered intracellular staining for NNT was the contrast between the background (diffuse neuropil staining) NNT immunoreactivity in the dementia cases versus the control cases, the latter being much lower.
Western Blotting
Western blots were performed both to evaluate the technical performance of the antibodies used for immunohistochemistry, and also to assess the immunoblotted bands that could be visualized for evidence of a high-molecular weight smear that is associated with pathogenic proteins in NDs. For the Western blots, antibodies were used that showed inclusion body-like structures on immunohistochemistry in Group 2 proteins as described above. Selected immunoblots are presented in Figures 7 and 8. Other Western blots performed are presented in Supplementary Data Figures S2–S15. For a list of cases used for Western blotting experiments in Supplementary Data Figures S2–S15, see Supplementary Data Table S3.
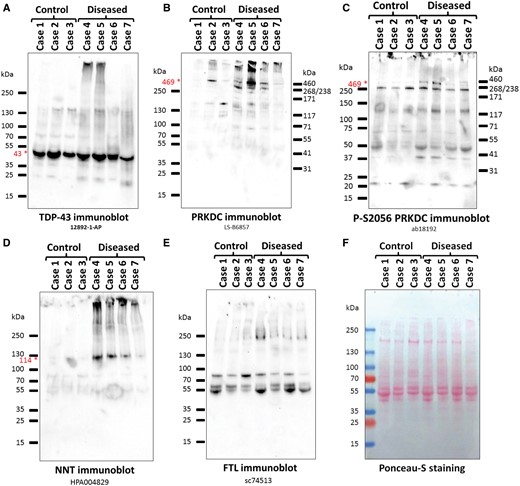
Western blots of detergent-insoluble, urea-soluble fractions from cases and controls, showing results of the samples used for mass spectrometry. Antibodies/antisera. (A) TDP-43 (12892-1-AP); (B) PRKDC (LS-B6857); (C) Phospho-S2056 PRKDC (ab18192); (D) NNT (HPA004829); (E) FTL (sc74513); (F) Ponceau-S staining as a loading control. All of the antibodies recognize proteins at the expected molecular weights (shown by red star and font), except for the FTL antibody that recognized a high-MW smear. The bands in the middle of the anti-FTL membrane correspond to the anti-mouse-HRP secondary antibody background (Supplementary Data Fig. S13).
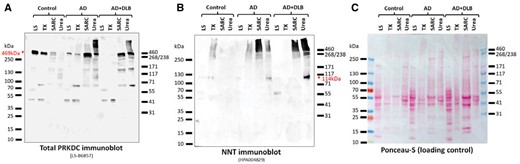
Western blots showing the enrichment of PRKDC and NNT in the detergent-insoluble protein fractions from a convenience sample of dementia cases and a control. These samples derive from a convenience sample comprising a control (age 81 at death, Braak NFT stage 0–i), AD (age 73, Braak NFT stage VI), and AD + DLB (age 86, Braak NFT stage V). The immunoblots show samples from the low salt (LS) fraction, Triton-X-100 fraction (TX), sarcosyl fraction (SARC), and the detergent-insoluble, urea-soluble fraction (Urea) for each case. The immunoblots were probed with the nonphosphorylated PRKDC antiserum (A; LS-B6857) and the NNT antiserum (B; HPA004829). For both PRKDC and NNT there is enrichment in the relatively insoluble fractions. (C) For loading control, the Ponceau-stained membrane is shown.
First, we tested whether the immunoblots for TDP-43 (a bona fide DIPP) could be correlated with expected results considering the known neuropathologic observations (Table 2). Overall, the TDP-43 immunoblot of the amygdalar urea-soluble fractions (Fig. 7A) showed suitable correlation with the preceding IHC studies of the corresponding Cases (Table 2) and our high molecular weight (high-MW) urea soluble fraction proteomics results (Table 3). We similarly tested for the presence of our Group 2 DIPP candidates with Western blotting of the urea-soluble fractions (Table 3) as summarized below.
PRKDC: Western blotting of the urea-soluble fractions used in the proteomics screen with an antiserum against total PRKDC detected bands at the expected size (469 kDa) (Fig. 7B). Additionally, a high-MW smear was also detected. Western blotting of the urea soluble fractions of 4 additional control and 6 additional AD cases with the total PRKDC antiserum detected stronger PRKDC signal in most AD cases than in the controls (Supplementary Data Fig. S2). Western blotting of the LS, TX, SARC, and urea fractions of control Case 11, AD (Case 12) and AD with dementia with Lewy body pathology (AD + DLB; Case 15) showed that whereas PRKDC was primarily present in the more soluble LS and TX fractions of the control, it significantly shifted towards the less soluble SARC and urea fractions in the AD cases (Fig. 8A). Western blotting with a phospho-S2056 PRKDC specific antiserum detected strong signal in dementia Cases 4 and 5 and weaker signal in Case 7 at the expected full-length size (Fig. 7C). Western blotting with the phospho-T2609 or phospho-T2647 PRKDC-specific antisera detected bands at lower molecular weights (Supplementary Data Figs. S4 and S5) that mostly corresponded to strong protein bands detected by Ponceau-S staining (Supplementary Data Fig. S15) or the background of the corresponding anti-rabbit-HRP secondary antibody control immunoblot (Supplementary Data Fig. S14).
NNT: Western blotting of the urea soluble fractions used in the proteomics screen with an anti-NNT antiserum detected the expected full-length NNT (114 kDa) in all dementia cases, but the corresponding band was barely detectable in the control cases (Fig. 7D). Western blotting of the LS, TX, SARC, and urea fractions of control Case 11 detected NNT as the expected 114 kDa band in the LS fraction, and in addition, as a high-MW smear in the TX fraction (Fig. 8B). In AD Case 12, NNT was also detected in the SARC and urea fractions, predominantly as a high-MW smear (Fig. 8B). In AD/DLB Case 15, an NNT-immunoreactive band at approximately 60 kDa was predominant in the LS fraction, NNT was almost undetectable in the TX fraction, and the NNT signal shifted towards the urea fraction even more than in AD Case 12 (Fig. 8B). Taken together, NNT fractionated into less soluble fractions significantly more in dementia cases than in controls.
FTL: Western blotting the urea-soluble fractions of the 3 control and 4 dementia cases used in proteomics with the anti-FTL antibody sc74513 did not reveal the presence of FTL at the expected full-length monomeric molecular weight (20 kDa). However, the antibody detected high-MW smears (Fig. 7E). The high-MW smear was strongest in Case 4, which correlated well with the mass spectrometric detection of FTL in Case 4 with a score of 66.3 (Table 3). Western blotting of the urea fractions of 4 additional control and 6 additional AD cases with the sc74513 FTL antibody revealed high-MW smears in AD Cases, as compared to the control cases (Supplementary Data Fig. S8). Well-defined bands are apparent in the sc74513 FTL blots in the 55–100 kDa range, likely representing secondary anti-mouse-HRP antibody background bands (Supplementary Data Fig. S13).
TNIK: Western blotting of the 7 urea soluble fractions used in the proteomics screen with 2 unrelated antibodies revealed a very similar pattern (Supplementary Data Fig. S9). The predominant band detected was approximately at the expected monomeric full-length size of TNIK (155 kDa), accompanied by a high-MW smear (Supplementary Data Fig. S9). Among dementia cases, the smear was strongest in Case 5.
AIFM1: Two different antibodies, LS-C353147 and ab32516 were used to probe the urea soluble fractions used in the proteomics screen. Prolonged exposure with sensitive ECL reagents detected bands in the 55–100 kDa range (Supplementary Data Figs. S11 and S12). However, the banding pattern was reminiscent of the corresponding anti-rabbit-HRP secondary antibody background (Supplementary Data Fig. S14), suggesting that the antibodies failed to detect specific AIFM1 signal in the urea fractions.
Genetic Association Study
We tested whether genes that encode Group 2 polypeptides (Table 3) had rare exonic polymorphisms that were associated with the phenotype of AD-type dementia. Analyzing whole exome sequence data obtained from ADSP, 3 rare gene variants were detected in NNT and 1 variant in each of TNIK and PRKDC (Table 5) with nominal significance (p < 0.05 uncorrected for multiple comparisons) with regard to association with AD-type dementia risk. A fourth NNT variant with p < 0.07 also is presented in Table 5. No rare variants in FTL were detected that showed association with AD dementia risk. In Table 5, the number of AD cases with the gene variant are the “Cases#”, and the number of persons with that gene variant in the entire sample (n = 10 488) are “Overall#.” Note that although the sample sizes were large in terms of numbers of cases and controls, the small numbers of rare variants in the cohorts led to small sample sizes for statistical comparisons in this context. Selected variants in NNT and PRKDC are nonsynonymous, and the rare variant rs192304760 in TNIK is located in a region exhibiting alternative splicing. One variant, rs139987446 in NNT, was present only in controls.
Results of Genetic Association Study Between Rare Coding Variants and Risk for Alzheimer-Type Dementia (Analyzed From ADSP, n = 5374 AD and 5094 Controls)
| Position* | rs ID* | OR* | Fisher p value | Permutation p value | Cases#/Overall# | Variant | MAF* | Alleles |
|---|---|---|---|---|---|---|---|---|
| NNT (Chr5p12) | ||||||||
| 43616091 | rs145205428 | 2.66 | 0.065 | 0.056 | 14/19 | Leu175Met | 0.000916 | A/C |
| 43655960 | rs139987446 | – | 0.028 | 0.012 | 0/5 | Arg693His | 0.000275 | A/G |
| 43675730 | rs139893206 | 2.37 | 0.018 | 0.011 | 25/35 | Ile918Val | 0.00165 | G/A |
| 43702785 | rs147457922 | 0.32 | 0.013 | 0.0077 | 7/24 | Pro1020Ala | 0.0011 | G/C |
| TNIK (Chr3q26) | ||||||||
| 170789119 | rs192304760 | 1.59 | 0.026 | 0.024 | 62/99 | c.3449-7A>T | 0.00477 | A/T |
| PRKDC (Chr8q11) | ||||||||
| 48711857 | rs8178225 | 1.37 | 0.0099 | 0.0091 | 166/280 | Gly3403Glu | 0.01354 | T/C |
| Position* | rs ID* | OR* | Fisher p value | Permutation p value | Cases#/Overall# | Variant | MAF* | Alleles |
|---|---|---|---|---|---|---|---|---|
| NNT (Chr5p12) | ||||||||
| 43616091 | rs145205428 | 2.66 | 0.065 | 0.056 | 14/19 | Leu175Met | 0.000916 | A/C |
| 43655960 | rs139987446 | – | 0.028 | 0.012 | 0/5 | Arg693His | 0.000275 | A/G |
| 43675730 | rs139893206 | 2.37 | 0.018 | 0.011 | 25/35 | Ile918Val | 0.00165 | G/A |
| 43702785 | rs147457922 | 0.32 | 0.013 | 0.0077 | 7/24 | Pro1020Ala | 0.0011 | G/C |
| TNIK (Chr3q26) | ||||||||
| 170789119 | rs192304760 | 1.59 | 0.026 | 0.024 | 62/99 | c.3449-7A>T | 0.00477 | A/T |
| PRKDC (Chr8q11) | ||||||||
| 48711857 | rs8178225 | 1.37 | 0.0099 | 0.0091 | 166/280 | Gly3403Glu | 0.01354 | T/C |
Position is referent to Chromosome build GRCh38/hg38; “rs ID”, identification of the coding variant; OR, odds ratio; MAF, minor allele frequency.
Results of Genetic Association Study Between Rare Coding Variants and Risk for Alzheimer-Type Dementia (Analyzed From ADSP, n = 5374 AD and 5094 Controls)
| Position* | rs ID* | OR* | Fisher p value | Permutation p value | Cases#/Overall# | Variant | MAF* | Alleles |
|---|---|---|---|---|---|---|---|---|
| NNT (Chr5p12) | ||||||||
| 43616091 | rs145205428 | 2.66 | 0.065 | 0.056 | 14/19 | Leu175Met | 0.000916 | A/C |
| 43655960 | rs139987446 | – | 0.028 | 0.012 | 0/5 | Arg693His | 0.000275 | A/G |
| 43675730 | rs139893206 | 2.37 | 0.018 | 0.011 | 25/35 | Ile918Val | 0.00165 | G/A |
| 43702785 | rs147457922 | 0.32 | 0.013 | 0.0077 | 7/24 | Pro1020Ala | 0.0011 | G/C |
| TNIK (Chr3q26) | ||||||||
| 170789119 | rs192304760 | 1.59 | 0.026 | 0.024 | 62/99 | c.3449-7A>T | 0.00477 | A/T |
| PRKDC (Chr8q11) | ||||||||
| 48711857 | rs8178225 | 1.37 | 0.0099 | 0.0091 | 166/280 | Gly3403Glu | 0.01354 | T/C |
| Position* | rs ID* | OR* | Fisher p value | Permutation p value | Cases#/Overall# | Variant | MAF* | Alleles |
|---|---|---|---|---|---|---|---|---|
| NNT (Chr5p12) | ||||||||
| 43616091 | rs145205428 | 2.66 | 0.065 | 0.056 | 14/19 | Leu175Met | 0.000916 | A/C |
| 43655960 | rs139987446 | – | 0.028 | 0.012 | 0/5 | Arg693His | 0.000275 | A/G |
| 43675730 | rs139893206 | 2.37 | 0.018 | 0.011 | 25/35 | Ile918Val | 0.00165 | G/A |
| 43702785 | rs147457922 | 0.32 | 0.013 | 0.0077 | 7/24 | Pro1020Ala | 0.0011 | G/C |
| TNIK (Chr3q26) | ||||||||
| 170789119 | rs192304760 | 1.59 | 0.026 | 0.024 | 62/99 | c.3449-7A>T | 0.00477 | A/T |
| PRKDC (Chr8q11) | ||||||||
| 48711857 | rs8178225 | 1.37 | 0.0099 | 0.0091 | 166/280 | Gly3403Glu | 0.01354 | T/C |
Position is referent to Chromosome build GRCh38/hg38; “rs ID”, identification of the coding variant; OR, odds ratio; MAF, minor allele frequency.
DISCUSSION
With the goal of identifying and characterizing novel DIPPs relevant to NDs, we extracted proteins from samples of human amygdalae and analyzed them with LC-ESI-MS/MS. Polypeptides present in detergent-insoluble extracts of patients with antemortem cognitive impairment were compared to those in control brains. On a subset of candidate DIPPs identified through these methods, immunohistochemical and Western blot experiments were performed, resulting in a refined list of novel candidate DIPPs: PRKDC, FTL, NNT, AIFM1, and TNIK. Finally, an in silico genetic association study was performed to detect rare variants of DIPP candidate genes which may be associated with an AD-type dementia phenotype.
There are limitations and caveats that apply to the current study. Although established pathologic markers are commonly identifiable in the amygdala at autopsy of older persons, these prior observations do not necessarily indicate that additional misfolded proteins (DIPPs other than Aβ, Tau, TDP-43, or α-synuclein) also can be discovered. In other words, researchers may have already identified all of the proteins that are pathologically deposited in the aged human amygdala in association with common dementia-inducing disorders. On the other hand, if many bona fide novel DIPPs were present in the samples we analyzed, we probably would not have detected them all. Additional limitations of the current study include the relatively small sample sizes, intrinsic imperfections in the proteomic analyses that do not include all peptides due to various biochemical factors, and, the potential pitfalls of false-negative and false-positive results when undertaking follow-up studies with commercially available antibodies. More tests will be required to evaluate these and other candidate DIPPs in greater detail—for example, investigations on ultrastructure, amyloidogenicity, and clinical-pathologic correlations that include analyses from other brain areas and specific subtypes of brain disease.
While the above caveats deserve consideration, the current study adds to an evolving appreciation of multiple misfolded proteins in the human brain. There have been previous analyses of the human amygdala proteome (48–50); however, no studies have been reported on the amygdala proteome in aged and demented subjects. Separate prior studies examined detergent-insoluble protein preparations from human brain samples outside of the amygdala, including mass spectrometric analyses of brain extracts from subjects with dementia-associated pathologies such as AD, CARTS, and frontotemporal lobar degeneration (15, 51–55). In the present study, as with prior published work, there was a tendency for cytoskeletal proteins including glial fibrillary acidic protein (GFAP), plectin, neurofilament, and microtubule-associated proteins to be detected in the detergent-insoluble fractions. Many of these protein species were represented in the samples from controls as well as dementia cases. There also have been other proteins previously identified to be enriched in the detergent-insoluble fraction of dementia subjects, including GFAP, STX1B, and APOE (15, 54, 55). Whereas the prior studies gained insights into the proteomics of human brain, there are aspects to our approach that were unique.
At least 5 facets of the study design and results are worthy of note: (1) Snap-frozen (PMI <4 hours) tissue from the amygdala was used from elderly persons with correlated clinical and pathologic data; (2) Proteins among the high-molecular weight portion of SDS gels were analyzed to maximize likelihood of identifying proteins with modifications that alter electrophoretic mobility; (3) Proteins were identified in the LC-MS/MS data analyses that would be expected to be present within inclusion bodies, including Tau, TDP-43, and α-synuclein; (4) Using Western blots and immunohistochemistry (following formic acid pretreatment), the study was oriented toward identifying candidate proteins that may be present in inclusion bodies; and (5) Protein-level studies were complemented with assessment of rare gene variants. It should be emphasized that we only tested a minority of the DIPP candidate proteins identified by proteomic analyses. Nonetheless, at least 4 proteins stood out as candidate DIPPs—PRKDC, FTL, NNT, and AIFM1 (leaving out TNIK for brevity’s sake)—and these are described in more detail below.
The protein kinase, DNA-activated, catalytic polypeptide (PRKDC) gene is located on human Chr. 8q11 and encodes a 4128 amino acid (∼469 kDa) polypeptide, which has also been termed DNA-PK, and DNA-PKcs. According to a study of laser-captured human cortical cells followed by RNA sequencing (Darmanis et al) (47), the PRKDC transcript is expressed in human brain at the 89th percentile for human brain transcripts, relatively evenly distributed across different human brain cell types. The PRKDC protein is a kinase that senses DNA damage and interacts with the Ku70/Ku80 protein to effect DNA double strand break repair and recombination (46), and is thought to be usually localized in the nucleus. A Prkdc null mutation is the causative genetic lesion in a mouse strain with the “SCID” (severe combined immunodeficiency) phenotype (56). There have been prior immunohistochemistry studies using antibodies against full-length PRKDC in human brains, including AD-type dementia (57, 58), and it has been suggested that PRKDC may contribute pathogenically to AD (59, 60), perhaps by phosphorylating Tau protein (61). It is possible that previous research showing decreased PRKDC expression and Ku protein binding in AD (62, 63) reflect that the detergent-soluble PRKDC protein pool decreases in parallel with the increase in the detergent-insoluble PRKDC (Fig. 8). LC-MS/MS data indicated the presence of many phosphorylated amino acid residues in PRKDC in cognitively impaired persons’ amygdalae (Supplementary Data Table S2). Correspondingly, immunohistochemistry using 3 different antisera against phosphorylated epitopes on PRKDC highlighted clear-cut inclusion bodies and thread-like structures in brains from cognitively impaired subjects. Based on these observations, either the immunohistochemical staining with these 3 separate antisera represent strong cross-reactions with other DIPPs, or else there is a large amount of PRKDC misfolded in brains of some demented persons. Consistent with the latter hypothesis, PRKDC immunoblots of proteins extracted from brains of individuals with antemortem cognitive impairment showed a high-molecular weight smear.
The Ferritin Light Chain (FTL) gene resides on Chr. 19q13 and encodes a 175 amino acid (∼20 kDa) protein. According to Darmanis et al (47), the FTL transcript is expressed in human brain at the 100th percentile for brain mRNAs, predominantly in macrophage/microglial cells and also highly expressed in astrocytes. This protein represents the small subunit of ferritin, an evolutionarily ancient protein complex which resides mostly in the cytoplasm and constitutes the major intracellular iron storage particle (64). Defects in the FTL gene are associated with a neurodegenerative disease, Neurodegeneration with Brain Iron Accumulation 3 (NBIA-3) (65). This autosomal dominant dystonia-chorea-dementia syndrome causes basal ganglia pathology and the NBIA/FTL mutation is predicted to lead to the formation of insoluble ferritin precipitates (66). NFT pathology has also been reported in NBIA-3 brains (67). Our samples presumably lack the rare NBIA-causing mutations, but in brains from demented subjects there were FTL-immunoreactive profiles, resembling inclusion bodies, and partial colocalization of FTL with TDP-43 pathology. On Western blots there was a high-molecular weight smear, that was increased in the impaired cases.
The Nicotinamide Nucleotide Transhydrogenase (NNT) gene is on Chr. 5p12, and it encodes a polypeptide of 1086 amino acids (∼114 kDa). According to Darmanis et al, the NNT transcript is expressed at the 88th percentile for human brain transcripts, evenly distributed in most brain cell types (47). The NNT gene product is an integral protein in the inner mitochondrial membrane, and this protein usually functions to promote the production of NADPH (68). In humans, NNT mutations cause primary adrenal insufficiency and are linked to oxidative stress (68, 69), whereas, in the mouse strain C57BL/6 J, which is used in many transgenic mice studies, the Nnt gene is mutated and the protein dysfunctional (70). In the present study, we only could identify a single commercially available antiserum that was suitable. Immunoblots with this antiserum against detergent-insoluble proteins from demented subjects revealed a conspicuous “smear” at high molecular weight, and general increase of background staining on immunohistochemistry, with scattered tangle-like structures in 5 out of 6 AD cases. It is also noted that NNT was absent in all 3 control cases and 2 out of 3 mild cognitive impairment cases (Table 4).
The apoptosis inducing factor mitochondria associated 1 (AIFM1) gene is on Chr. Xq26 and codes for a polypeptide of 613 amino acids (∼67 kDa). The protein has also been given the designations PDCD8 and AIF. According to Darmanis et al, the AIFM1 transcript is expressed at the 85th percentile in human brain, relatively enriched in “mature astrocytes” in terms of cell-specific expression (47). This protein is an NADH oxidoreductase and flavoprotein, normally found in the mitochondrial intermembrane space (71). In certain conditions, AIFM1 gets transported to the nucleus where it is a DNA-binding protein that participates in processes of apoptosis, potentiating chromosomal fragmentation and, ultimately, nuclear disassembly (71). Mutations in AIFM1 can cause Cowchock syndrome (72), a disease characterized by axonal defects and clinical cognitive impairment, and AIFM1 mutations can also cause severe mitochondrial encephalomyopathy (73). In the current study, AIFM1 immunohistochemistry showed structures that resembled inclusion bodies in brains of dementia subjects, in contrast to staining of control brains which was negligible. In our hands, the AIFM1 antibodies did not work on immunoblots of urea-soluble proteins.
In conclusion, we report new candidate DIPPs identified from the proteome of the amygdala of cognitively impaired research subjects. The results of our studies may indicate prevalent phenomena, since candidate DIPPs were present in multiple cases in a small convenience sample. We used commercially available antibodies for immunohistochemical studies, and provided detailed protocols, to enable researchers in other laboratories to perform similar studies. Additional experiments are required to determine if pathologic markers referent to the newly identified candidate DIPPs (e.g., PRKDC, FTL, NNT, AIFM1, and a larger set of proteins we did not test further) are associated with cognitive impairment in older persons’ brains independently of other neuropathologic changes. If such associations do in fact exist, they may help to explain the data from different centers which indicate that not all the variance in cognition or nerve cell loss in the human brain are explained by known pathological markers (22, 74–76). Given the evolving awareness of the misfolded proteome, it is possible that the polypeptide products of a rather large number of different genes may be misfolded and may participate in disease-driving processes in the brains of elderly persons.
ACKNOWLEDGMENTS
We are sincerely grateful for the research volunteers and clinical colleagues at the University of Kentucky Alzheimer’s Disease Center. Thanks to Dr. Adam Bachstetter for methodological advice and reagents, and to Drs. Peter Davies, Virginia Lee, and Eddie Lee for antibodies. The ADSP Acknowledgement is provided in the Supplementary Data.
REFERENCES
Author notes
The study was supported by NIH grants P30 AG028383, R01 AG042419, R01 AG042475, T32 AG 000242, R21 NS095299 (to J.G.), and VA MERIT award I01 BX002149 (to H.Z.). The Orbitrap mass spectrometer was acquired by NIH Grant S10 RR029127 (to H.Z.).
The authors have no duality or conflicts of interest to declare.
Supplementary Data can be found at http://www.jnen.oxfordjournals.org.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}